Multiple myeloma from basic Robbins and Levinson
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Searching Images
multiple myeloma plasma cells bone marrow pathology
Loading Image
Loading Image
Here is a comprehensive summary of Multiple Myeloma as covered in Robbins & Kumar Basic Pathology:
Multiple Myeloma
— Robbins & Kumar Basic Pathology (Robbins Pathology)
Overview
Multiple myeloma is a malignant proliferation of plasma cells derived from a single clone. It is one of the most common lymphoid malignancies — approximately 30,000 new cases/year in the United States. Key epidemiological points:
- Median age at diagnosis: 70 years
- More common in males
- Occurs more frequently in people of African descent in the US (reason unknown)
- Principally involves the bone marrow and is associated with lytic skeletal lesions
M Protein Production
The neoplastic plasma cells secrete a monoclonal immunoglobulin (M protein):
- IgG — 60% (most common)
- IgA — 20–25%
- IgM, IgD, IgE — rare
- Remaining cases: only κ or λ free light chains (Bence Jones proteins)
Pathogenesis
| Mechanism | Details |
|---|---|
| Chromosomal translocations | Fuse the IgH locus (chr 14) to oncogenes such as cyclin D1 and cyclin D3 → dysregulation of D cyclins → increased cell proliferation |
| IL-6 | Produced by bone marrow stromal fibroblasts and macrophages; supports myeloma cell proliferation |
| MYC translocations | Seen late in disease, associated with aggressive behavior |
Pathologic Effects (Morbidity & Mortality)
1. Skeletal Destruction
- Myeloma-derived factors upregulate RANKL on stromal cells → activates osteoclasts
- Tumor factors also inhibit osteoblast function
- Net result: increased bone resorption → hypercalcemia and pathologic fractures
2. Immune Defects
- Myeloma cells compromise normal B-cell function through uncertain mechanisms
- Despite elevated serum immunoglobulin (M protein), production of functional antibodies is profoundly depressed
- Result: high risk for bacterial infections
3. Renal Dysfunction ("Myeloma Kidney")
Multiple mechanisms acting alone or in combination:
- Obstructive proteinaceous casts in distal convoluted tubules and collecting ducts (mostly Bence Jones proteins + albumin + tubular secretions)
- Light chain deposition in glomeruli/interstitium — as AL amyloid or linear deposits
- Hypercalcemia → dehydration, renal stones
- Increased incidence of bacterial pyelonephritis (from hypogammaglobulinemia)
Morphology
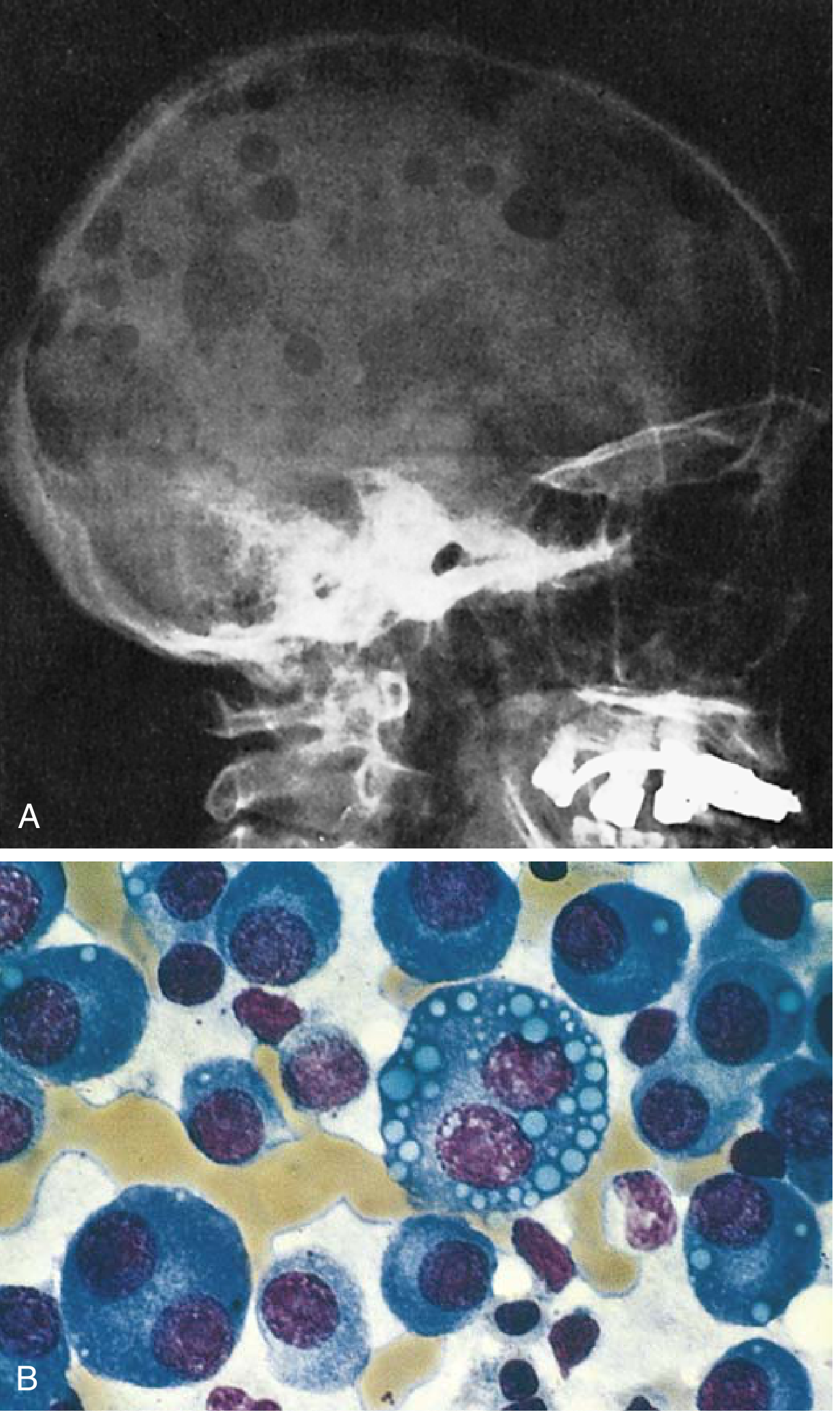
FIG. 10.28 — Robbins & Kumar. (A) Punched-out lytic lesions in the skull calvaria. (B) Myeloma cells in marrow — note prominent nucleoli and Russell bodies (cytoplasmic Ig inclusions).
Skeletal lesions:
- Multifocal destructive lytic lesions involving vertebral column, ribs, skull, pelvis, femur, clavicle, scapula
- Arise in medullary cavity → erode cancellous bone → destroy cortex
- Appear as "punched-out" defects 1–4 cm in diameter on imaging (Fig. 10.28A)
- Pathologic fractures most frequent in vertebral column and femur
Bone marrow:
- Plasma cells typically >30% of marrow cellularity (required for diagnosis)
- Cells may resemble normal plasma cells or show abnormal features:
- Prominent nucleoli
- Russell bodies — cytoplasmic inclusions containing immunoglobulin (Fig. 10.28B)
- Late disease: spread to viscera and soft tissues; terminal stage may show a leukemic picture
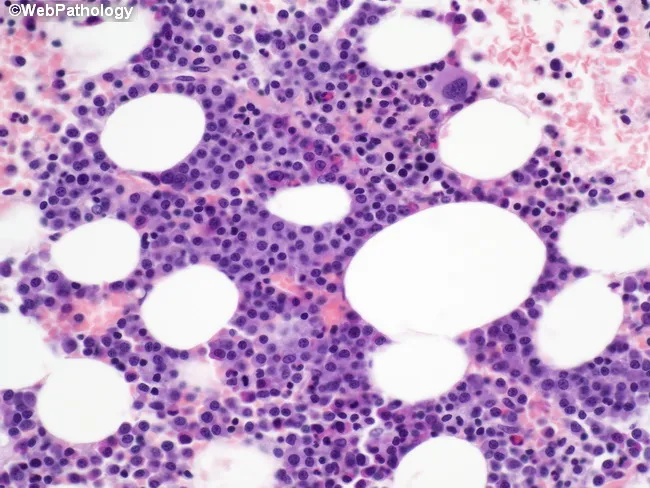
H&E bone marrow biopsy — sheets of malignant plasma cells replacing normal marrow architecture.
Renal (myeloma kidney):
- Proteinaceous Bence Jones casts in distal tubules and collecting ducts
- Casts surrounded by multinucleate giant cells (macrophage-derived)
- Adjacent tubular epithelium undergoes necrosis/atrophy from Bence Jones protein toxicity
- Other renal changes: metastatic calcification, AL amyloidosis (glomeruli/vessel walls), bacterial pyelonephritis, and rarely neoplastic plasma cell infiltrates in the interstitium
Laboratory Findings
| Finding | Detail |
|---|---|
| Serum M protein | Detectable in most cases |
| Bence Jones protein (urine) | Free light chains |
| Both M protein + free light chains | 60–70% of cases |
| Free light chains only | ~20% of cases |
| Nonsecretory myeloma | ~1% — absence of M component does NOT exclude diagnosis |
| CBC | Normocytic normochromic anemia; may have leukopenia and thrombocytopenia (marrow replacement) |
Clinical Features
Clinical manifestations stem from four main processes:
- Plasma cell effects on skeleton — pathologic fractures, chronic bone pain
- Excessive/abnormal immunoglobulins — hyperviscosity (with IgM/IgA particularly), amyloidosis
- Suppression of humoral immunity — recurrent bacterial infections
- Renal insufficiency — occurs in up to 50% of patients; second leading cause of death (after infections); correlated with degree of Bence Jones proteinuria
Hypercalcemia → neurologic symptoms: confusion, weakness, lethargy
Diagnosis
- Imaging: typical punched-out lytic bone lesions
- Labs: M protein + Bence Jones protein quantification
- Definitive: bone marrow examination (plasma cells >30%)
Prognosis & Treatment
| Aspect | Detail |
|---|---|
| Untreated (multiple bony lesions) | Survival rarely >6–12 months |
| Smoldering myeloma | May not require treatment for years |
| Median survival | ~5 years |
| Cure | Not yet achieved |
Therapeutic options:
- Proteasome inhibitors (e.g., bortezomib) — myeloma cells accumulate misfolded Ig chains; proteasome inhibition exacerbates this, triggering apoptosis
- Thalidomide-like drugs (IMiDs) — stimulate degradation of pro-oncogenic proteins in myeloma cells
- Bisphosphonates — inhibit osteoclast-mediated bone resorption; reduce fractures and hypercalcemia
- Hematopoietic stem cell transplantation — prolongs life but not curative
- CAR-T cell therapy — cytotoxic T cells engineered to recognize plasma cell antigens (newer approach)
Note: There is no "Levinson" textbook in the available library. The content above is fully sourced from Robbins & Kumar Basic Pathology — the standard Robbins text for basic pathology. If you meant a different book (e.g., Jawetz, Melnick & Adelberg's Medical Microbiology by Levinson), please clarify and I'll look it up.
Non Hodgkin's lymphoma from basic Robbins and Levinson
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
I now have all the content needed for a comprehensive answer. Let me compile it.
Non-Hodgkin Lymphoma (NHL)
— Robbins & Kumar Basic Pathology (Robbins Pathology)
Overview
Non-Hodgkin lymphomas (NHLs) are a heterogeneous group of lymphoid malignancies. The majority (~85–90%) are of B-cell origin; the remainder are T-cell or NK-cell tumors. Key general features:
- Most arise in lymph nodes but extranodal presentations are common
- NHL is broadly divided into indolent (slow-growing, often incurable) and aggressive (fast-growing, but often curable) types
- Painless lymphadenopathy is the most common presentation
The major NHL subtypes covered in Robbins Basic Pathology are outlined below.
1. Follicular Lymphoma
Frequency: ~30% of adult NHLs in the United States (less common in Asian populations)
Pathogenesis
-
85% have a (14;18) translocation — fuses the BCL2 gene (chr 18) to the IgH locus (chr 14)
- Results in overexpression of BCL2 protein → inhibition of apoptosis → cell survival
- Additional mutations in histone-modifying genes suggest epigenetic contributions
Morphology
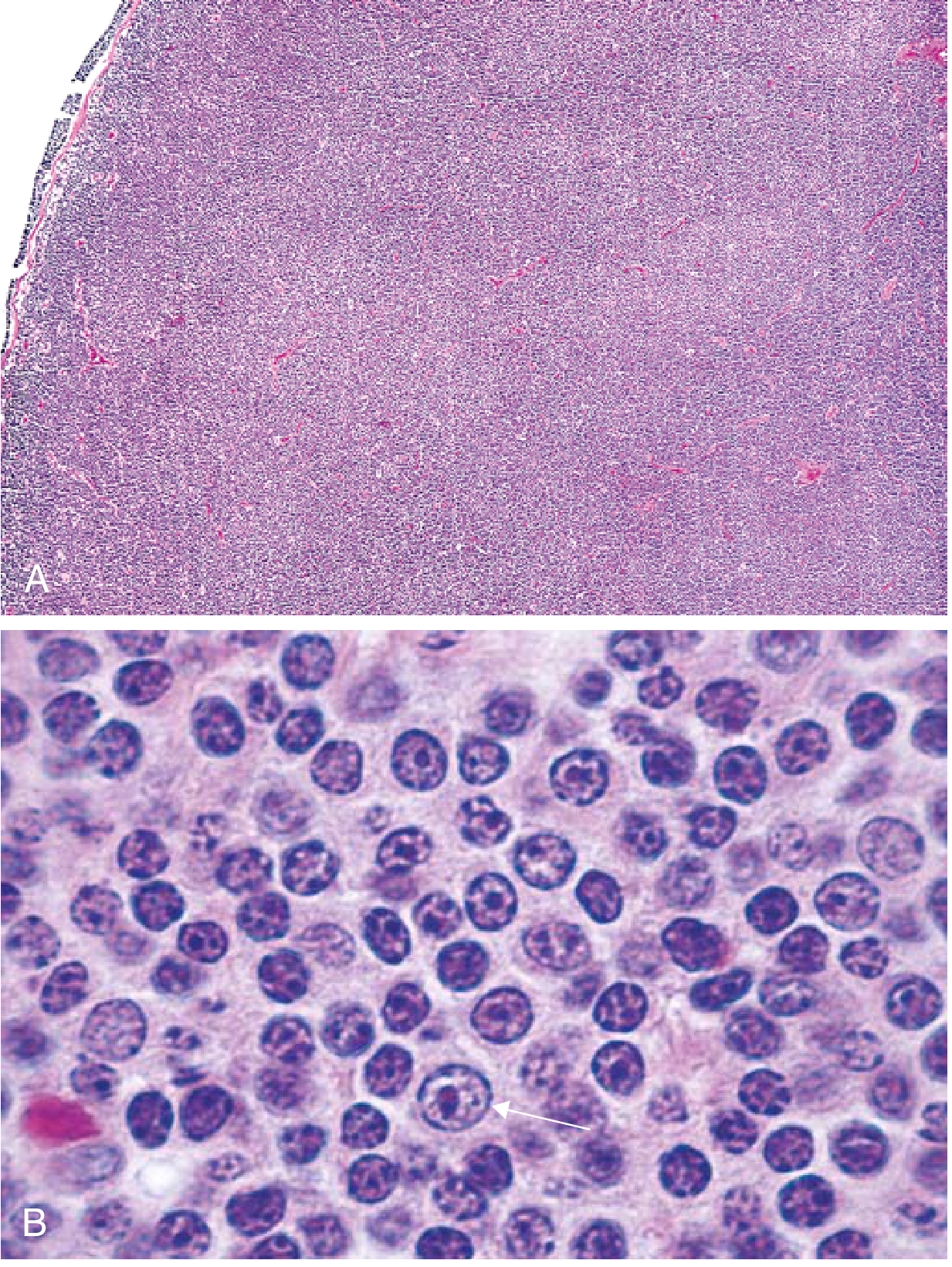
- Lymph nodes effaced by a distinctly nodular proliferation
- Predominantly centrocytes — slightly larger than resting lymphocytes with angular "cleaved" nuclei, coarse chromatin, indistinct nucleoli
- Mixed with variable centroblasts — larger cells with vesicular chromatin, several nucleoli
- Few mitoses, no apoptosis — distinguishes from follicular hyperplasia (where both are prominent)
- Occasional predominance of large cells = more aggressive histology
Immunophenotype
| Marker | Status |
|---|---|
| CD20 | + |
| CD10 | + (germinal center marker) |
| BCL6 | + (germinal center transcription factor) |
| BCL2 protein | Overexpressed (distinguishes from reactive follicles) |
Clinical Features
- Adults >50 years; equal sex distribution
- Presents as painless, generalized lymphadenopathy
- Bone marrow involved at diagnosis in ~80% of cases
- Indolent but incurable — median survival ~10 years
- Transformation to aggressive DLBCL can occur (worsens prognosis)
2. Diffuse Large B-Cell Lymphoma (DLBCL)
Frequency: Most common adult NHL — ~35% of all adult NHLs
Pathogenesis
| Alteration | Frequency | Effect |
|---|---|---|
| BCL6 gene rearrangement (3q27) | ~1/3 | Overexpression of BCL6 → germinal center B-cell dysregulation |
| BCL6 promoter mutations | Even higher fraction | Same effect |
| (14;18) BCL2 translocation | ~30% | Anti-apoptotic; some may be transformed follicular lymphomas |
| MYC translocations | Subset | Pro-proliferative |
Morphology
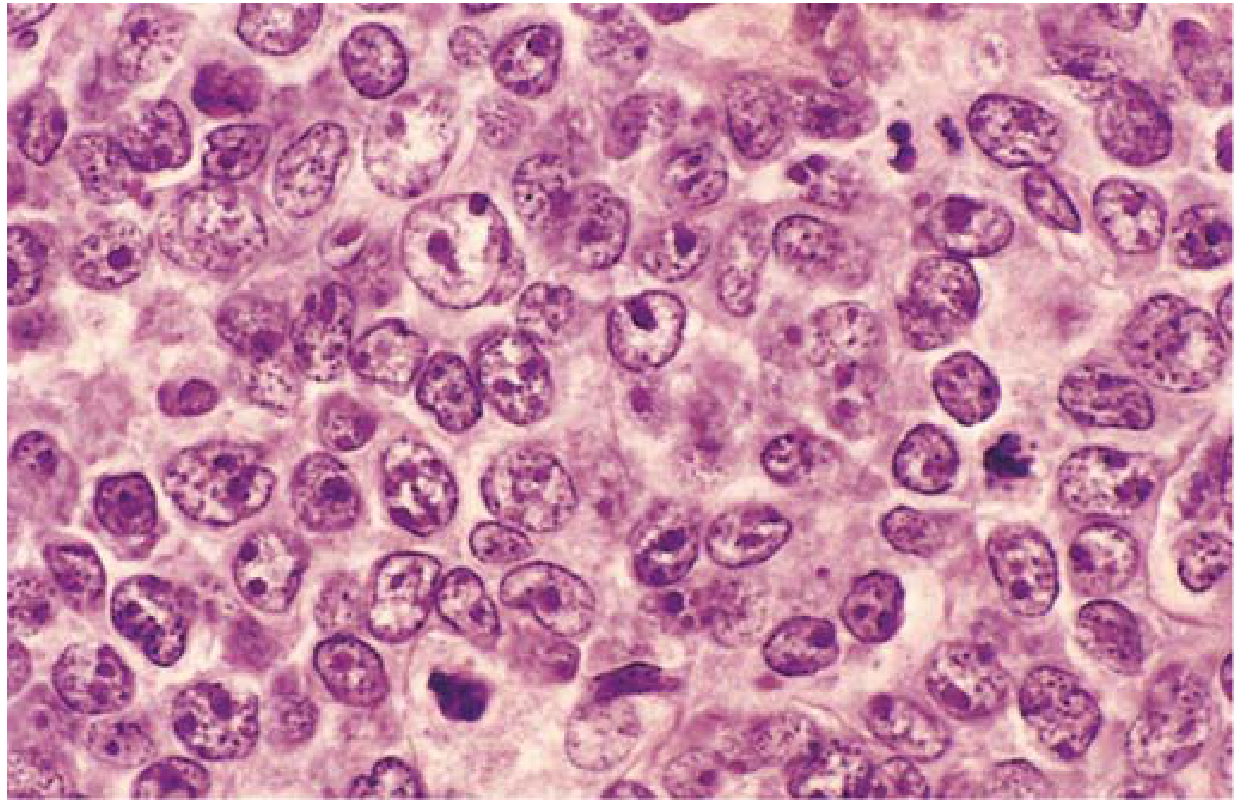
- Large cells (3–4× resting lymphocyte size), variable morphology
- Round/oval nuclei with dispersed chromatin, several nucleoli, pale cytoplasm
- Some tumors have multilobate vesicular nuclei with prominent central nucleoli
- Occasionally highly anaplastic with Reed-Sternberg–like giant cells
Immunophenotype
- CD20 positive; variably express surface IgM/IgG, CD10, MYC, BCL2
Special Subtypes
- EBV-associated DLBCL — arises in AIDS, iatrogenic immunosuppression, elderly; posttransplant cases may be polyclonal initially and regress with restored immunity
- Primary effusion lymphoma — associated with HHV8/KSHV; arises in pleural, pericardial, or peritoneal cavities; HHV8 encodes oncoproteins including a cyclin D1 homolog
- Mediastinal large B-cell lymphoma — often involves thymus; young women; spreads to abdomen and CNS; frequently shows PD-L1 overexpression (immune evasion mechanism)
Clinical Features
- Median age ~60 years, but can occur at any age (~15% of childhood lymphomas)
- Rapidly enlarging symptomatic mass at one or more sites
- Extranodal presentations common — GI tract most frequent extranodal site
- Liver, spleen, and bone marrow involvement uncommon at diagnosis
- Aggressive and rapidly fatal if untreated
- With intensive chemotherapy + anti-CD20 immunotherapy (rituximab): complete remission in 60–80%; ~50% appear cured
- CAR-T cell therapy (anti-CD19) can be curative for relapsed/refractory cases
3. Mantle Cell Lymphoma
Frequency: ~6% of all NHLs; mainly in men >50 years
Pathogenesis
- (11;14) translocation — fuses cyclin D1 gene to the IgH locus
- Overexpression of cyclin D1 → promotes G1→S cell cycle progression (via Rb hyperphosphorylation) → uncontrolled proliferation
Morphology
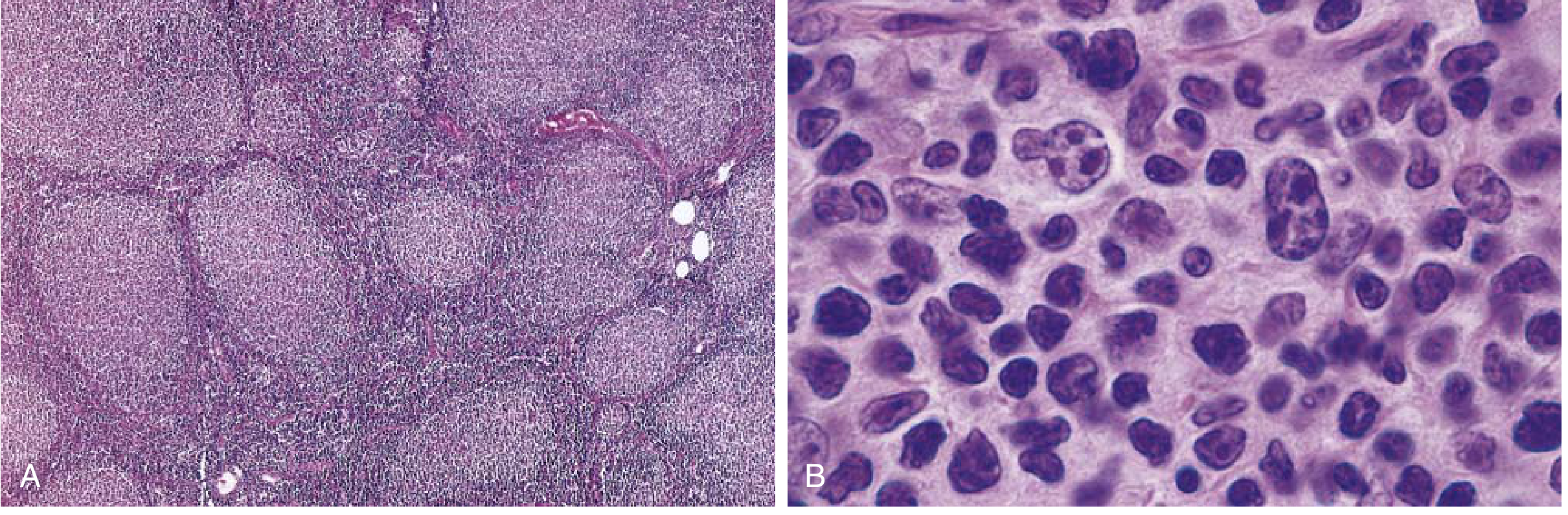
- Diffuse or vaguely nodular pattern in lymph nodes
- No proliferation centers (distinguishes from CLL/SLL)
- Cells slightly larger than normal lymphocytes; irregular nucleus, inconspicuous nucleoli, scant cytoplasm
- Occasional blastoid variant (larger, lymphoblast-like cells)
- Bone marrow involved in most cases; peripheral blood in ~20%
- GI involvement: lymphomatoid polyposis (multifocal submucosal nodules resembling polyps)
Immunophenotype
- Surface IgM, IgD; CD20+; CD5+ (shared with CLL/SLL); Cyclin D1+ (distinguishing marker)
Clinical Features
- Fatigue, lymphadenopathy, generalized disease (bone marrow, spleen, liver, GI tract)
- Moderately aggressive, incurable
- Median survival: 6–7 years
- Treatment includes BTK inhibitors (tumor cells depend on B-cell receptor signaling for survival)
4. Extranodal Marginal Zone Lymphoma (MALToma)
Origin: Indolent B-cell tumor arising in epithelial tissues — stomach, salivary glands, bowel, lungs, orbit, breast
Pathogenesis
- Arises within sites of chronic inflammation:
- Autoimmune: salivary glands (Sjögren syndrome), thyroid (Hashimoto thyroiditis)
- Infection: H. pylori gastritis (most common)
- Tumor cells depend on inflammatory cytokines from H. pylori–specific T cells for growth
- Key insight: H. pylori eradication with antibiotics often causes tumor regression — a unique cancer therapy
5. Burkitt Lymphoma
Frequency: ~30% of childhood NHLs in the United States; endemic in parts of Africa
Pathogenesis
- Translocations involving MYC gene (chromosome 8):
- Most common: t(8;14) — MYC fused to IgH locus (chr 14)
- Variants: t(8;2) and t(8;22) — MYC fused to κ or λ light chain loci
- MYC = master regulator of Warburg metabolism (aerobic glycolysis) → drives rapid cell growth
- Burkitt lymphoma is among the fastest growing human tumors
- EBV association: most endemic (African) cases + ~20% of sporadic cases
Morphology
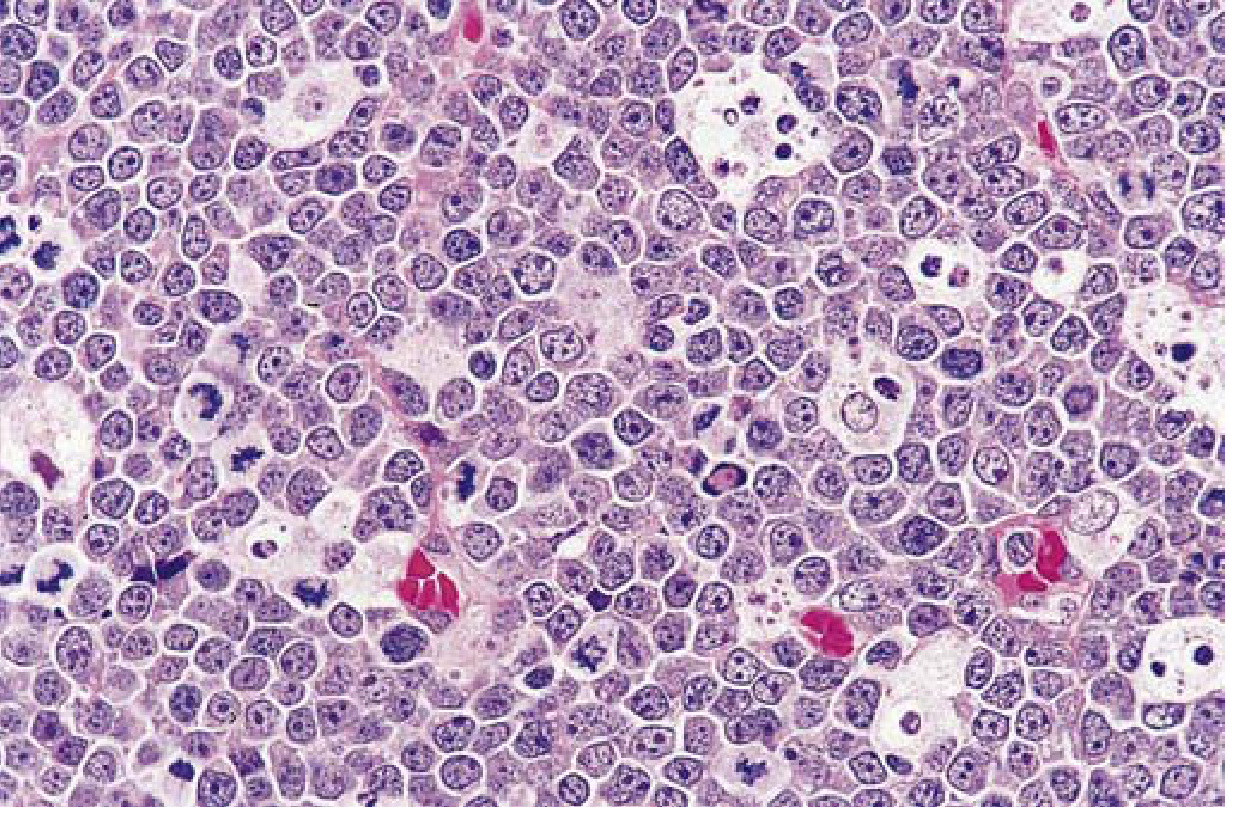
- Intermediate-sized tumor cells with round/oval nuclei, 2–5 distinct nucleoli
- Moderate basophilic/amphophilic cytoplasm with lipid vacuoles (on smears)
- Very high proliferation AND apoptosis rates → tissue macrophages ingesting nuclear debris → "starry sky" pattern (macrophages = stars; dark tumor cells = sky)
Immunophenotype
- Surface IgM; CD20+; CD10+; BCL6+ (germinal center B-cell markers)
Clinical Features
- Affects children and young adults
- Usually arises at extranodal sites:
- Endemic (African): maxillary/mandibular jaw masses
- Sporadic (North America): abdominal tumors — bowel, retroperitoneum, ovaries
- Leukemic presentations: must distinguish from B-ALL (different treatment)
- Highly aggressive but majority cured with very intensive chemotherapy
6. Other Notable NHL Entities
Hairy Cell Leukemia
- Uncommon, indolent B-cell neoplasm
- Nearly all cases have activating BRAF mutations (also mutated in melanoma, colon cancer)
- Morphology: fine hairlike cytoplasmic projections
- Markers: CD20+, surface Ig+, CD11c+, CD103+ (distinctive)
- Treated with purine analogues (e.g., cladribine) — induces prolonged remissions
Peripheral T-Cell Lymphomas
- Uncommon, heterogeneous group
- Most are aggressive with a poor prognosis
- Example: Anaplastic large cell lymphoma (ALCL) — characterized by ALK kinase fusions (e.g., t(2;5) → NPM-ALK); responds to ALK inhibitors
Comparison Summary Table
| Feature | Follicular | DLBCL | Mantle Cell | Burkitt | MALToma |
|---|---|---|---|---|---|
| Frequency (adult NHL) | ~30% | ~35% | ~6% | Common in children | Variable |
| Key translocation | t(14;18) BCL2 | BCL6 rearrangement | t(11;14) cyclin D1 | t(8;14) MYC | Variable |
| Behavior | Indolent | Aggressive | Moderately aggressive | Highly aggressive | Indolent |
| Curability | No | Yes (~50%) | No | Yes (intensive chemo) | Yes (H. pylori Rx) |
| Key marker | BCL2+ | CD20+ | Cyclin D1+, CD5+ | CD10+, BCL6+ | — |
| "Starry sky" | No | No | No | Yes | No |
Note on "Levinson": No textbook by Levinson exists in the available library. The content above is entirely from Robbins & Kumar Basic Pathology. If you meant a specific microbiology or immunology text, please clarify the full title.
This is a shared conversation. Sign in to Orris to start your own chat.