Why nsaid is contraindicated in Bronchial asthma?
Reading File
Finding Sources
Finding Sources
Searching PubMed
NSAID aspirin exacerbated respiratory disease asthma mechanism
Reading File
Reading File
Reading File
Loading Image
Reading File
Excellent - I now have all the detail needed for a thorough answer, including the mechanism diagram. Here is a complete explanation:
Why NSAIDs are Contraindicated in Bronchial Asthma
The core issue is a condition called Aspirin-Exacerbated Respiratory Disease (AERD), which occurs in approximately 5-20% of asthmatics (higher rates - up to 20% - in patients with severe asthma, particularly women, and those with nasal polyps).
The Central Mechanism: COX-1 Inhibition and Arachidonic Acid Shunting
Arachidonic acid (AA) released from cell membranes can be metabolized via two competing pathways:
- COX pathway - produces prostaglandins (PGE2, PGH2, PGD2) and thromboxane
- Lipoxygenase (5-LO) pathway - produces leukotrienes (LTC4, LTD4, LTE4)
Under normal conditions, PGE2 acts as a brake on the 5-LO pathway - it inhibits 5-lipoxygenase activity and suppresses leukotriene production.
When NSAIDs inhibit COX-1:
- PGE2 production falls sharply
- The inhibitory brake on 5-lipoxygenase is removed
- Arachidonic acid is shunted into the leukotriene pathway
- Massive overproduction of cysteinyl leukotrienes (LTC4, LTD4, LTE4) results
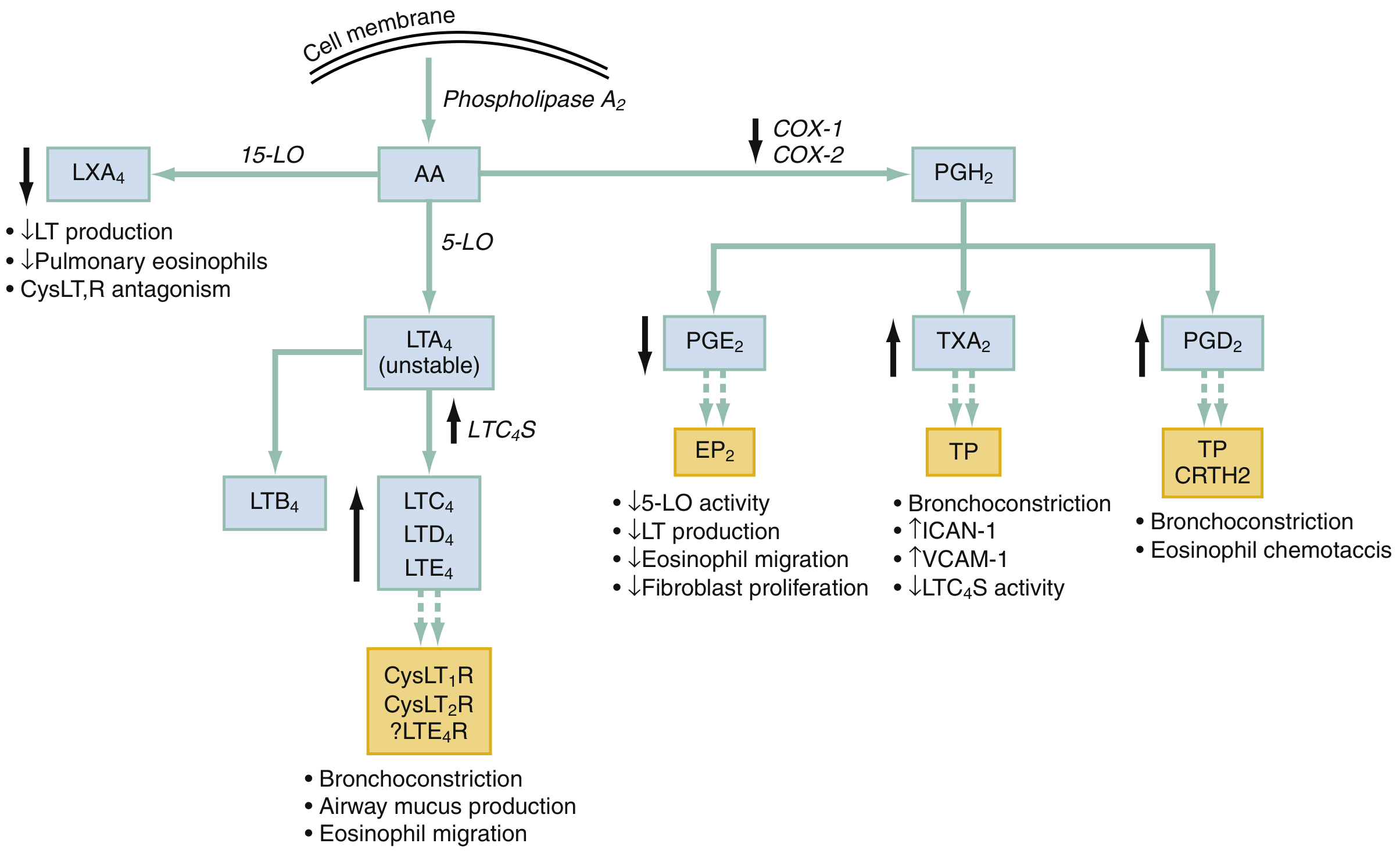
Fig. 59.5 from Rosen's Emergency Medicine: COX inhibition decreases PGE2, removing its inhibitory effect on 5-LO, resulting in increased LTC4/LTD4/LTE4 and bronchoconstriction.
Consequences of Excess Leukotrienes
Cysteinyl leukotrienes binding to CysLT1R and CysLT2R cause:
- Bronchoconstriction (potent - far more so than histamine)
- Airway mucus hypersecretion
- Mucosal edema
- Eosinophil migration into airway tissue
- Increased bronchial hyperreactivity
This is a non-IgE-mediated (non-allergic) pharmacological reaction - it is not an allergy to the drug itself, but a direct consequence of enzyme inhibition.
Additional Pathway Disruptions
The diagram also shows that COX inhibition causes:
- Decreased LXA4 (lipoxin A4) - a pro-resolution mediator that normally suppresses leukotriene production and reduces pulmonary eosinophilia
- Increased TXA2 activity (relative) - causes bronchoconstriction and upregulates ICAM-1/VCAM-1
- Increased PGD2 signaling via CRTH2 receptor - causes bronchoconstriction and eosinophil chemotaxis
All these changes compound the bronchoconstriction.
Clinical Features of AERD (Samter's Triad)
The classic clinical tetrad includes:
- Asthma
- Nasal polyps
- Eosinophilic sinusitis
- Sensitivity to COX-1 inhibiting drugs (aspirin, NSAIDs)
Ingestion of even a small aspirin dose causes profound bronchoconstriction, nasal congestion, flushing, and abdominal cramping - sometimes within minutes to 1-2 hours. AERD accounts for 25% of asthmatics requiring mechanical ventilation.
Selective COX-2 Inhibitors are Generally Safe
Because this reaction is specifically triggered by COX-1 inhibition, patients with AERD generally tolerate selective COX-2 inhibitors (e.g., celecoxib). This is why paracetamol (acetaminophen) at normal doses is also usually safe - it is a very weak COX-1 inhibitor.
Cross-Reactivity
All NSAIDs that inhibit COX-1 show universal cross-reactivity in AERD patients. There is no chemical structural similarity required - the pharmacological effect is what matters.
Management Considerations
- Avoid all COX-1 inhibiting NSAIDs in known asthmatics, especially those with nasal polyps
- Inhaled corticosteroids (ICS) are the primary therapy
- Leukotriene antagonists (montelukast, zafirlukast) and 5-LO inhibitors (zileuton) are particularly useful in AERD - they directly block the aberrant pathway
- Aspirin desensitization is a specialized protocol that can paradoxically reduce long-term AERD severity
Sources: Rosen's Emergency Medicine, 10th Ed. (p. 932); Katzung's Basic & Clinical Pharmacology, 16th Ed. (p. 559); Goldman-Cecil Medicine, 2-vol. set
This is a shared conversation. Sign in to Orris to start your own chat.