Tell me blood coagulation pathway and all things involved in it and tell me about various disease that cause bleeding disorders and it correlation clinically to Ent
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Searching Images
coagulation cascade intrinsic extrinsic pathway factors diagram
Reading File
Loading Image
Here is a comprehensive breakdown of the blood coagulation pathway, the diseases causing bleeding disorders, and their ENT clinical correlations.
Blood Coagulation — Complete Overview
1. Overview of Hemostasis
Hemostasis — the process that arrests bleeding after vascular injury — proceeds in four sequential phases:
- Vascular spasm — immediate smooth muscle contraction triggered by myogenic reflex, local autacoids, and thromboxane A2 from platelets
- Platelet plug formation — primary hemostasis
- Coagulation cascade — secondary hemostasis, resulting in a fibrin clot
- Fibrous tissue repair — permanent sealing of the vessel
2. Platelet Plug (Primary Hemostasis)
Platelets (thrombocytes, 150,000–450,000/μL, lifespan 8–12 days, formed from megakaryocytes) become activated when vascular injury exposes subendothelial collagen:
- Platelet adhesion — via von Willebrand factor (vWF) bridging collagen to platelet glycoprotein Ib (GP Ib)
- Platelet activation — release of ADP, thromboxane A2, serotonin → vasoconstriction + further platelet recruitment
- Platelet aggregation — GP IIb/IIIa receptors cross-link platelets via fibrinogen bridges
- Platelet factor 3 (phospholipid membrane) — provides the surface for clotting reactions
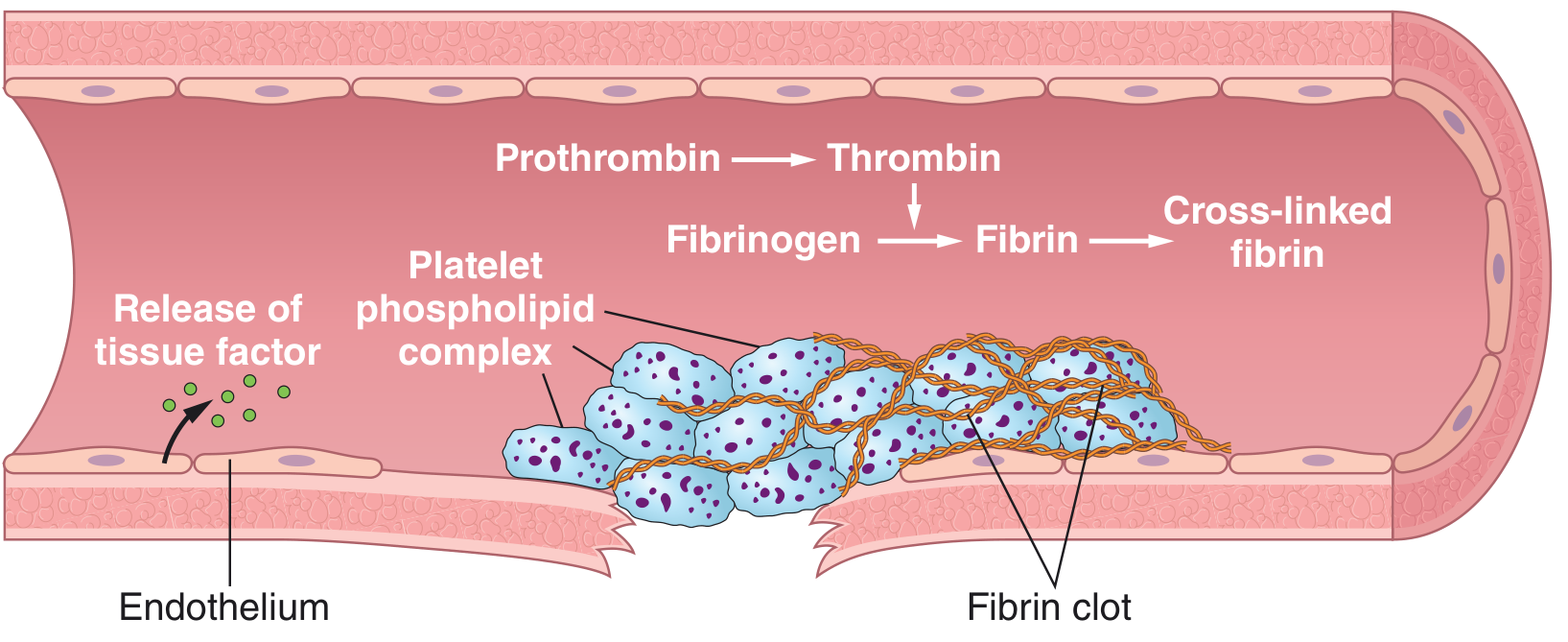
3. Coagulation Cascade (Secondary Hemostasis)
The cascade is a series of zymogen → active enzyme reactions, each requiring a membrane-bound complex of: enzyme + substrate + cofactor + Ca²⁺. This confines clot formation to the injury site.
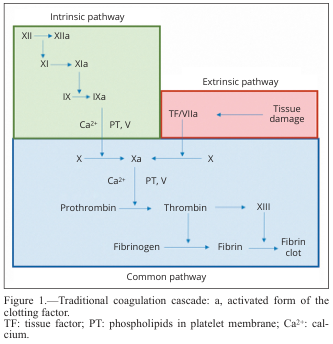
A. Extrinsic Pathway (Initiation Phase)
Triggered by tissue factor (TF / Factor III) exposed after vascular injury:
| Step | Event |
|---|---|
| Injury | Subendothelial TF exposed |
| TF + Factor VIIa | Form a complex on phospholipid surface |
| TF/VIIa complex | Activates Factor X → Xa AND Factor IX → IXa |
This is the initiation phase. Small amounts of thrombin are generated here, but the pathway is quickly inhibited by TFPI (Tissue Factor Pathway Inhibitor).
B. Intrinsic Pathway (Amplification Phase)
Classically initiated by contact activation; physiologically acts to amplify thrombin generation:
| Step | Factor Activated | Cofactors |
|---|---|---|
| Contact with collagen/foreign surface | XII → XIIa (+ HMWK, prekallikrein) | — |
| XIIa acts on | XI → XIa | HMWK |
| XIa acts on | IX → IXa | Ca²⁺ |
| IXa + VIIIa + Ca²⁺ + PL | Tenase complex → X → Xa | Factor VIII (antihemophilic factor) |
Factor VIII deficiency = Hemophilia A | Factor IX deficiency = Hemophilia B
C. Common Pathway (Final Phase)
Both pathways converge here:
| Step | Complex / Action |
|---|---|
| Xa + Va + Ca²⁺ + PL | Prothrombinase complex |
| Prothrombin (II) → Thrombin (IIa) | Central enzyme of coagulation |
| Thrombin cleaves fibrinogen | Fibrinopeptides A & B removed → Fibrin monomers |
| Fibrin monomers polymerize | Form fibrin strands (weak, H-bond linked) |
| Factor XIII → XIIIa (activated by thrombin) | Cross-links fibrin → insoluble, stable clot |
Positive Feedback Loops
- Thrombin activates Factor V, Factor VIII, and Factor XIII — all amplify further clot formation
- Thrombin activates Factor XI → further amplification via intrinsic pathway
Role of Calcium (Ca²⁺)
Ca²⁺ is required for every step except the first two (XII activation and XI activation). This is why chelating agents (citrate, oxalate, EDTA) prevent coagulation in stored blood.
Role of Vitamin K
Required by the liver for γ-carboxylation of glutamic acid residues on Factors II (prothrombin), VII, IX, X, and Protein C/S. Without this modification, these factors cannot bind Ca²⁺ and are nonfunctional.
4. Clot Dissolution (Fibrinolysis)
Once healing occurs, clot is dissolved by:
- Plasmin (from plasminogen, activated by t-PA and u-PA)
- Plasmin cleaves fibrin → fibrin degradation products (FDPs/D-dimers)
- Regulated by PAI-1 and α₂-antiplasmin
5. Natural Anticoagulants (Control Mechanisms)
| Inhibitor | Mechanism |
|---|---|
| Antithrombin III | Inhibits thrombin and Xa, IXa, XIa (heparin potentiates this) |
| Protein C + S | Inactivate factors Va and VIIIa |
| TFPI | Inhibits TF/VIIa/Xa complex |
| Thrombomodulin | Endothelial receptor; thrombin bound to it activates Protein C |
| Prostacyclin (PGI₂) | Inhibits platelet aggregation |
6. Coagulation Factors Reference Table
| Factor | Name | Pathway | Synthesized | Notes |
|---|---|---|---|---|
| I | Fibrinogen | Common | Liver | Substrate for clot |
| II | Prothrombin | Common | Liver (Vit K) | Cleaved to thrombin |
| III | Tissue factor | Extrinsic | Subendothelial | Initiates cascade |
| IV | Calcium (Ca²⁺) | All | — | Essential cofactor |
| V | Labile factor | Common | Liver | Cofactor in prothrombinase |
| VII | Proconvertin | Extrinsic | Liver (Vit K) | Shortest half-life |
| VIII | Antihemophilic factor | Intrinsic | Liver/endothelium | Deficient in Hemophilia A |
| IX | Christmas factor | Intrinsic | Liver (Vit K) | Deficient in Hemophilia B |
| X | Stuart-Prower factor | Common | Liver (Vit K) | Central convergence point |
| XI | Plasma thromboplastin antecedent | Intrinsic | Liver | |
| XII | Hageman factor | Intrinsic | Liver | Contact activation |
| XIII | Fibrin stabilizing factor | Common | Liver/platelets | Cross-links fibrin |
| vWF | von Willebrand factor | — | Endothelium | Carrier for VIII; platelet adhesion |
7. Diseases Causing Bleeding Disorders
A. Platelet / Primary Hemostasis Disorders
| Disorder | Defect | Bleeding Pattern |
|---|---|---|
| Thrombocytopenia | Low platelet count (<150,000/μL) | Petechiae, purpura, mucosal bleeding, epistaxis |
| ITP (Immune Thrombocytopenic Purpura) | Autoantibodies destroy platelets | Petechiae, mucosal bleeding |
| Bernard-Soulier syndrome | Absent GP Ib → no vWF binding | Prolonged bleeding time |
| Glanzmann thrombasthenia | Absent GP IIb/IIIa | Failure of aggregation |
| Aspirin/NSAID use | COX inhibition → ↓TXA₂ → ↓aggregation | Mucosal bleeding, ↑surgical blood loss |
| Uremia | Qualitative platelet dysfunction | Mucosal bleeding |
| Myeloproliferative disorders | Abnormal platelet function | Variable |
B. Coagulation Factor Deficiencies (Secondary Hemostasis Disorders)
| Disorder | Factor Deficient | Pathway Affected | Inheritance | Lab |
|---|---|---|---|---|
| Hemophilia A | Factor VIII | Intrinsic | X-linked recessive | ↑aPTT, normal PT |
| Hemophilia B (Christmas disease) | Factor IX | Intrinsic | X-linked recessive | ↑aPTT, normal PT |
| Hemophilia C | Factor XI | Intrinsic | Autosomal | ↑aPTT |
| von Willebrand disease (vWD) | vWF (± VIII) | Primary + Intrinsic | Autosomal dominant (Type 1/2), recessive (Type 3) | ↑BT, ↑aPTT |
| Vitamin K deficiency | II, VII, IX, X | All | Acquired | ↑PT + ↑aPTT |
| Liver disease | Multiple factors | All | Acquired | ↑PT, ↑aPTT, ↓fibrinogen |
| DIC (Disseminated Intravascular Coagulation) | Consumption of all factors | All | Acquired | ↑PT, ↑aPTT, ↑D-dimer, ↓platelets |
| Afibrinogenemia / Dysfibrinogenemia | Fibrinogen (Factor I) | Common | Rare | ↑TT |
| Factor XIII deficiency | Cross-linking enzyme | Common | Rare | Normal PT/aPTT, clot dissolves in urea |
C. Vascular Disorders
| Disorder | Mechanism | ENT Relevance |
|---|---|---|
| Hereditary Hemorrhagic Telangiectasia (HHT / Osler-Weber-Rendu) | Vascular malformations | Severe, recurrent epistaxis — hallmark ENT presentation |
| Ehlers-Danlos syndrome | Connective tissue fragility | Mucosal bleeding, easy bruising |
| Scurvy (Vit C deficiency) | ↓Collagen synthesis → vessel wall weakness | Gingival bleeding |
| Henoch-Schönlein Purpura | IgA vasculitis | Purpura, GI bleeding |
8. Laboratory Tests of Coagulation
| Test | Measures | Normal | Prolonged In |
|---|---|---|---|
| PT (INR) | Extrinsic + common | ~12 sec / INR 1.0 | Factor VII deficiency, Warfarin, liver disease, Vit K deficiency |
| aPTT | Intrinsic + common | 25–35 sec | Hemophilia A/B/C, Heparin, lupus anticoagulant |
| Bleeding time | Platelet function | 1–6 min | Thrombocytopenia, vWD, aspirin |
| Thrombin time (TT) | Fibrinogen → Fibrin | 10–12 sec | Afibrinogenemia, heparin, DIC |
| D-dimer | Fibrinolysis/fibrin degradation | <0.5 μg/mL | DIC, PE, DVT |
| Platelet count | Number | 150–450 × 10³/μL | Any thrombocytopenia |
9. ENT Clinical Correlations of Bleeding Disorders
Epistaxis (Nosebleed) — The Most Common ENT Bleeding Presentation
Epistaxis is the sentinel ENT symptom of nearly all bleeding disorders. It arises from the highly vascular Kiesselbach's plexus (Little's area) on the anterior nasal septum.
| Disorder | ENT / Clinical Correlation |
|---|---|
| von Willebrand Disease | Most common inherited cause of mucosal bleeding including epistaxis; recurrent epistaxis, especially in children and adolescents; heavy menstrual bleeding (HMB) in women |
| Hemophilia A/B | Less commonly presents with epistaxis, but post-tonsillectomy/adenoidectomy (T&A) hemorrhage is a serious risk; delayed bleeding (hours to days) after surgery is characteristic |
| Thrombocytopenia (ITP, drug-induced) | Petechiae on palate and buccal mucosa; epistaxis; gingival bleeding; ↑risk of post-operative hemorrhage |
| HHT (Osler-Weber-Rendu) | Recurrent, bilateral, severe epistaxis — often the presenting symptom; telangiectasias visible on nasal mucosa, lips, tongue; may require laser cautery, septodermoplasty, or embolization |
| Liver disease / Cirrhosis | Multiple factor deficiency → ↑surgical and post-T&A bleeding; also low platelet count due to hypersplenism |
| Vitamin K deficiency | Deficiency of II, VII, IX, X → prolonged PT → increased operative bleeding in ENT procedures |
| Aspirin / NSAID / Anticoagulant use | Among the most common causes of refractory epistaxis; must be identified pre-operatively |
| DIC | Mucosal oozing from multiple ENT sites; seen in sepsis, trauma — serious systemic emergency |
| Leukemia / Bone marrow failure | Thrombocytopenia → petechiae on palate, gingival bleeding, epistaxis; oral cavity often involved |
| Uremia | Platelet dysfunction → mucosal bleeding, troublesome epistaxis unresponsive to local measures; treated with DDAVP |
Pre-operative ENT Assessment
Patients undergoing tonsillectomy, adenoidectomy, functional endoscopic sinus surgery (FESS), septoplasty, or rhinoplasty must be evaluated for:
- History of prior excessive bleeding (tonsillectomy is a classic challenge)
- Family history of bleeding
- Drug history (aspirin, warfarin, NOACs, NSAIDs, herbal supplements)
- Screening labs: PT, aPTT, platelet count
Patterns That Guide Diagnosis
| Clinical Pattern | Likely Disorder |
|---|---|
| Petechiae + mucosal bleeding + epistaxis | Platelet disorder (thrombocytopenia, vWD) |
| Deep tissue hematoma + post-op delayed bleeding | Coagulation factor deficiency (Hemophilia) |
| Recurrent bilateral epistaxis + family history + telangiectasias | HHT |
| Epistaxis in child + heavy periods in mother | von Willebrand disease |
| Post-tonsillectomy bleeding 7–10 days later | Secondary hemorrhage (infection, eschar separation — but factor deficiency must be excluded) |
| Epistaxis + prolonged PT + ↑aPTT + ↓platelets + elevated D-dimer | DIC (sepsis, trauma, malignancy) |
Key Summary
| Pathway | Key Factors | Lab Test | Major Disease |
|---|---|---|---|
| Extrinsic | TF, VII | PT / INR | Warfarin, Vit K deficiency, liver disease |
| Intrinsic | XII, XI, IX, VIII | aPTT | Hemophilia A (VIII), Hemophilia B (IX) |
| Common | X, V, II, I, XIII | Both PT + aPTT | DIC, liver failure, afibrinogenemia |
| Platelet | GP Ib, GP IIb/IIIa, vWF | Bleeding time / PFA-100 | vWD, ITP, Glanzmann, Bernard-Soulier |
Sources: Guyton and Hall Textbook of Medical Physiology; Miller's Anesthesia 10e; Goldman-Cecil Medicine International Edition
This is a shared conversation. Sign in to Orris to start your own chat.