
Solve with reason
Reading File
Finding Sources
Answer: C) Polycythemia Vera
Clinical Reasoning
Every feature in this case maps precisely to Polycythemia Vera (PV), a myeloproliferative neoplasm driven by a JAK2 mutation causing unregulated overproduction of red blood cells (and often platelets and white cells).
| Finding in Case | Why It Points to PV |
|---|---|
| Headache, blurred vision | Hyperviscosity from elevated RBC mass reduces cerebral blood flow |
| Prior stroke | Thrombotic complication — hallmark of PV due to hyperviscosity and thrombocytosis |
| Fatigue | Constitutional symptom of myeloproliferative disease |
| Episodic epistaxis | Paradoxical bleeding from acquired von Willebrand disease & platelet dysfunction |
| Generalized itching (pruritus) | Especially post-bath/shower — classic "aquagenic pruritus" from histamine release by mast cells; highly specific for PV |
| Splenomegaly (8 cm) | Extramedullary hematopoiesis, sequestration of excess blood cells |
| Hypertension (BP 170/90) | Increased blood viscosity and volume |
Why Not the Others?
- A) Essential thrombocythemia — Causes thrombosis and splenomegaly, but aquagenic pruritus is rare, and the primary abnormality is platelets, not red cells. Hypertension and stroke are less characteristic.
- B) Secondary polycythemia — Reactive erythrocytosis (from hypoxia, EPO-secreting tumors, etc.) — does not cause splenomegaly, pruritus, or leukocytosis/thrombocytosis; JAK2 mutation absent.
- D) Chronic myeloid leukemia (CML) — BCR-ABL driven; causes leukocytosis and splenomegaly, but not polycythemia or aquagenic pruritus; stroke is not a typical presentation.
Key Diagnostic Markers for PV (WHO Criteria)
Major criteria:
- Elevated hemoglobin/hematocrit (Hb >16.5 g/dL men)
- Hypercellular bone marrow with trilineage growth (panmyelosis)
- JAK2 V617F mutation (or JAK2 exon 12 mutation)
Minor criterion: Subnormal serum erythropoietin level
Diagnosis requires all 3 major, or 2 major + minor criterion.
— Rosen's Emergency Medicine; Goldman-Cecil Medicine; Bradley & Daroff's Neurology in Clinical Practice
Essential Thrombocytopenia
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Here is a comprehensive overview of Essential Thrombocythemia (ET) — also called Essential Thrombocytosis or Essential Thrombocytemia.
Note on terminology: "Essential Thrombocytopenia" is not a recognized entity. You likely mean Essential Thrombocythemia (ET) — a condition of excess platelets (thrombocytosis/thrombocythemia), not decreased platelets (thrombocytopenia). These are opposite conditions.
Essential Thrombocythemia (ET)
Definition
ET is a clonal myeloproliferative neoplasm (MPN) arising from a hematopoietic stem cell, characterized by autonomous overproduction of platelets (>450,000/μL) without a definable external cause.
Pathogenesis & Molecular Mutations
ET results from mutations that constitutively activate JAK-STAT signaling, mimicking continuous growth factor stimulation. Over 90% of cases harbor one of three driver mutations:
| Mutation | Frequency | Mechanism |
|---|---|---|
| JAK2 V617F | ~50–60% | Constitutive JAK2 kinase activation |
| CALR (calreticulin) | ~30% | Mutant CALR is secreted → binds & activates thrombopoietin receptor (MPL) — an oncogenic autocrine loop |
| MPL | ~5–10% | Activating mutation of the thrombopoietin receptor itself |
| Mutation-negative | ~10% | Hereditary forms or uncommon MPL variants |
The same JAK2 V617F mutation is found in Polycythemia Vera — why some patients develop ET vs. PV is not fully understood. Iron deficiency can sometimes mask PV, making it appear as ET.
Epidemiology
- Incidence: 1–3 per 100,000/year
- Usually occurs past age 60, but can occur in young adults
- Female predominance (unlike primary myelofibrosis or reactive thrombocytosis)
Clinical Features
Symptoms
- Up to 50% are asymptomatic at diagnosis (incidental thrombocytosis on CBC)
- Vasomotor symptoms: headache, lightheadedness, visual disturbances (amaurosis fugax, scintillating scotomata), syncope, chest pain
- Erythromelalgia: burning, throbbing pain of hands/feet with erythema and warmth — caused by occlusion of small arterioles by platelet aggregates
- Thrombosis: DVT, pulmonary embolism, stroke/TIA, MI, portal/hepatic/mesenteric vein thrombosis, cerebral venous sinus thrombosis
- Hemorrhage: paradoxically, when platelet count >1.5 million/μL, acquired von Willebrand disease develops — large vWF multimers are absorbed and destroyed by the platelet mass → bleeding risk
Signs
- Splenomegaly: mild, in ~10–50% of patients (extramedullary hematopoiesis)
- Abnormally large platelets on peripheral smear
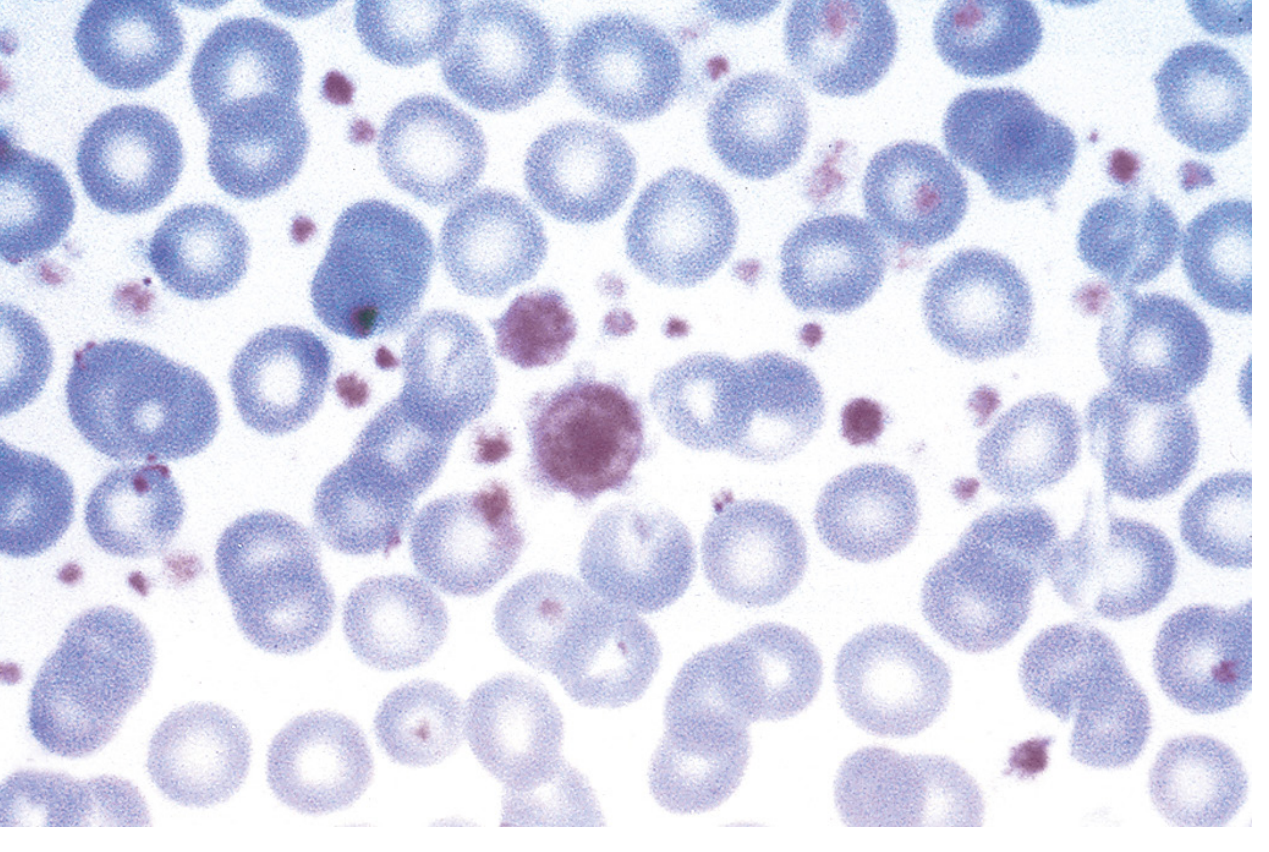
Bone Marrow Morphology
- Mildly increased cellularity
- Markedly increased megakaryocytes, including abnormally large forms
- Delicate reticulin fibrils (but no overt fibrosis — distinguishes from primary myelofibrosis)
- Mild extramedullary hematopoiesis
WHO Diagnostic Criteria (ET requires all 4 major OR first 3 major + minor)
Major Criteria:
- Platelet count ≥ 450,000/μL
- Bone marrow biopsy showing megakaryocyte proliferation with large mature forms; no significant increase in granulopoiesis or erythropoiesis; no relevant fibrosis
- Does not meet criteria for CML, PV, PMF, MDS, or other myeloid neoplasm
- Presence of JAK2, CALR, or MPL mutation
Minor Criterion:
- Presence of a clonal marker OR absence of evidence for reactive thrombocytosis
Distinguishing ET from Other Causes of Thrombocytosis
| Feature | ET | Reactive Thrombocytosis |
|---|---|---|
| Driver mutation | JAK2/CALR/MPL present | Absent |
| Splenomegaly | Possible | Absent |
| Platelet morphology | Giant, dysfunctional platelets | Normal |
| Thrombotic risk | Elevated | Not elevated |
| Causes | Clonal MPN | Infection, inflammation, iron deficiency, hemorrhage, post-splenectomy |
Complications
- Thrombosis (arterial > venous): most likely in patients with high platelet counts and homozygous JAK2 mutations
- Acquired von Willebrand disease (platelet count >1.5 million/μL) → bleeding
- Rare progression to: acute myeloid leukemia (AML) or myelofibrosis
Treatment
| Situation | Management |
|---|---|
| Asymptomatic, no CV risk factors | Observation only — no therapy needed |
| Platelet count >1 million/μL + bleeding | Avoid aspirin (worsens bleeding); use tranexamic acid or ε-aminocaproic acid |
| Symptomatic / high thrombotic risk | Cytoreduction: Hydroxyurea (first-line), Pegylated IFN-α, or Anagrelide |
| Prevention of TIA | Hydroxyurea + aspirin > Anagrelide + aspirin |
| Venous thrombosis prevention | Anagrelide + aspirin may be superior |
Key principle: Normalizing the platelet count does not reliably prevent arterial or venous thrombosis. Avoid alkylating agents and ³²P — they increase leukemic transformation risk, especially combined with hydroxyurea.
Prognosis
- Survival is not significantly different from the general population
- Median survival: 12–15 years
- ET is considered more benign than previously thought; progression to AML is more often a consequence of therapy than the disease itself
— Harrison's Principles of Internal Medicine 22E; Goldman-Cecil Medicine; Robbins & Cotran Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.