Idiopathic pulmonary fibrosis in detail
Reading File
search_medical_text
idiopathic pulmonary fibrosis etiology pathophysiology clinical features diagnosis
search_medical_text
idiopathic pulmonary fibrosis treatment management antifibrotic prognosis
search_medical_image
idiopathic pulmonary fibrosis HRCT usual interstitial pneumonia UIP pattern
GradeResponse
GradeResponse
GradeResponse
Here is a comprehensive overview of idiopathic pulmonary fibrosis (IPF):
Idiopathic Pulmonary Fibrosis (IPF)
Definition
IPF is a chronic, progressive, fibrosing interstitial pneumonia of unknown cause, associated with the radiological and histopathological pattern of Usual Interstitial Pneumonia (UIP). It occurs primarily in older adults and carries a poor prognosis. (ATS/ERS Guidelines, p. 2)
Epidemiology
| Parameter | Detail |
|---|---|
| Age of onset | Typically >60 years (rare <50) |
| Sex | Male predominance (~2:1) |
| Prevalence | ~13–20 per 100,000 (higher in older populations) |
| Incidence | Increasing globally |
Risk factors:
- Cigarette smoking (strongest environmental risk; ~75% of patients are current or ex-smokers)
- Occupational dust exposures (wood, metal, agricultural dust)
- Gastroesophageal reflux disease (GERD)
- Viral infections (EBV, CMV, HHV-8)
- Genetic predisposition — MUC5B promoter variant (rs35705950) is the most common genetic risk factor; also TERT, TERC, SFTPC, SFTPA2 mutations (telomere/surfactant biology)
- Family history (familial IPF = ~5% of cases)
Pathophysiology
The prevailing model is one of aberrant wound healing rather than primary inflammation:
- Repetitive alveolar epithelial injury → apoptosis of type II pneumocytes in genetically susceptible individuals
- Dysregulated repair → abnormal activation and proliferation of fibroblasts/myofibroblasts
- Fibroblastic foci form — hallmark histological feature — with excessive deposition of extracellular matrix (collagen)
- Key mediators:
- TGF-β1 (master profibrotic cytokine)
- PDGF, VEGF, FGF
- Epithelial-mesenchymal transition (EMT)
- Oxidative stress, telomere shortening, senescence
- Progressive architectural distortion → honeycombing → respiratory failure
The inflammatory cascade is now considered secondary, not causal — explaining why anti-inflammatory therapies fail.
Clinical Features
Symptoms
- Progressive exertional dyspnea (insidious onset, months–years)
- Dry, persistent cough (often refractory)
- Fatigue, weight loss (late)
- Rare: pleuritic chest pain
Signs
- Bibasal fine inspiratory ("Velcro") crackles — hallmark
- Clubbing (~50%)
- Cyanosis and signs of pulmonary hypertension (right heart failure) in advanced disease
Diagnosis
Diagnosis requires multidisciplinary discussion (MDD) involving pulmonologist, radiologist, and pathologist. Key steps:
Step 1: Exclude Other Causes of ILD
- Connective tissue disease (CTD-ILD): SLE, RA, SSc, PM/DM
- Hypersensitivity pneumonitis (HP)
- Drug-induced ILD (amiodarone, methotrexate)
- Occupational/environmental ILD
- Sarcoidosis
Step 2: High-Resolution CT (HRCT) — Pattern Classification
HRCT is the cornerstone of diagnosis. The UIP pattern is classified as:
| HRCT Category | Features | Surgical Biopsy Needed? |
|---|---|---|
| Typical UIP | Bilateral, subpleural, basal-predominant; honeycombing ± traction bronchiectasis; no features against UIP | No |
| Probable UIP | Reticular pattern, traction bronchiectasis, no honeycombing; subpleural, basal | May need biopsy |
| Indeterminate for UIP | Subtle reticulation; may have features atypical for UIP | Often needs biopsy |
| Alternative diagnosis | Features suggesting another ILD | Biopsy or alternative workup |
HRCT — Typical UIP Pattern
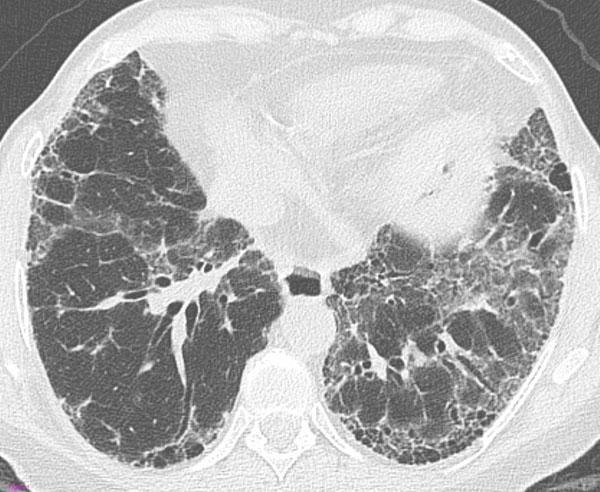
Axial HRCT demonstrating the "definite UIP" pattern: bilateral subpleural honeycombing (clustered thick-walled cysts), traction bronchiectasis within fibrotic areas, and a peripheral reticular pattern with basal predominance — classic for IPF.
Step 3: Histopathology (when needed)
Surgical lung biopsy (VATS) or transbronchial lung cryobiopsy (TBLC) — TBLC now conditionally recommended as a less invasive alternative.
UIP histologic features:
- Fibroblastic foci (key feature)
- Patchy fibrosis with temporal heterogeneity (old + new fibrosis coexisting)
- Honeycombing (subpleural, basal)
- Minimal inflammation
Step 4: Additional Diagnostics
| Test | Purpose |
|---|---|
| PFTs | Restrictive pattern: ↓TLC, ↓FVC, ↓DLCO, normal or ↑FEV1/FVC |
| 6-Minute Walk Test (6MWT) | Exercise capacity, baseline and monitoring |
| BAL (bronchoalveolar lavage) | Exclude HP, eosinophilia, infection |
| Serologies (ANA, RF, anti-CCP, anti-Jo-1, etc.) | Exclude CTD-ILD |
| Genomic classifier testing | Peripheral blood test (Envisia) to distinguish UIP vs non-UIP — newly addressed in 2022 guidelines |
| Echocardiography | Screen for pulmonary hypertension |
| Sleep study | Screen for OSA (common comorbidity) |
Differential Diagnosis
| Condition | Key Distinguishing Feature |
|---|---|
| HP (chronic) | Upper/mid-lobe predominance, centrilobular nodules, exposure history |
| NSIP | Bilateral GGO, subpleural sparing, younger age, CTD association |
| Cryptogenic organizing pneumonia | Consolidation, peribronchovascular, responds to steroids |
| Sarcoidosis | Upper lobe, lymphadenopathy, non-caseating granulomas |
| Asbestosis | Pleural plaques, occupational history |
| CTD-ILD (RA, SSc) | Serological markers, systemic features |
Management
(ATS/ERS Guidelines, p. 18)
Pharmacological Treatment
Antifibrotic Agents (Disease-Modifying)
| Drug | Mechanism | Dose | Key Side Effects |
|---|---|---|---|
| Pirfenidone (Esbriet) | Anti-inflammatory, antifibrotic, antioxidant; inhibits TGF-β | 2403 mg/day (tid) | Nausea, rash, photosensitivity, ↑LFTs |
| Nintedanib (Ofev) | Tyrosine kinase inhibitor (PDGFR, FGFR, VEGFR) | 150 mg bid | Diarrhea, nausea, ↑LFTs, teratogenic |
Both drugs reduce FVC decline by ~50% and reduce acute exacerbation risk. They do not reverse fibrosis but slow progression. Both are conditionally recommended by ATS/ERS guidelines; choice is individualized based on comorbidities and tolerability.
Not recommended: Corticosteroids (alone or combined with azathioprine + N-acetylcysteine), anticoagulation (warfarin), sildenafil for ILD-related dyspnea, and imatinib.
Non-Pharmacological Management
| Intervention | Indication/Detail |
|---|---|
| Long-term oxygen therapy | Resting hypoxemia (SpO₂ <88%) or exertional desaturation |
| Pulmonary rehabilitation | Improves exercise tolerance, dyspnea, QoL |
| Lung transplantation | Definitive treatment; single or bilateral; refer early (at diagnosis if high-risk features); 5-year survival ~50% |
| Antacid therapy (PPIs/H2 blockers) | GERD treatment recommended (GERD highly prevalent; micro-aspiration may drive injury) |
| Palliative care | Symptom management (cough, dyspnea, anxiety, depression); discuss early |
Comorbidity Management
| Comorbidity | Approach |
|---|---|
| Pulmonary hypertension | Evaluate with echo/RHC; consider targeted therapy |
| OSA | CPAP; very common in IPF |
| Lung cancer | IPF is an independent risk factor; screen per guidelines |
| GERD | Treat aggressively |
| Coronary artery disease | Common in same demographic |
Acute Exacerbations of IPF (AE-IPF)
- Definition: Acute worsening with new bilateral GGO/consolidation on HRCT (beyond background UIP) with no identifiable cause
- Triggers: Infection, aspiration, surgery, bronchoscopy — or idiopathic
- Incidence: ~5–10%/year
- Management:
- High-dose corticosteroids (empiric, evidence limited)
- Broad-spectrum antibiotics (to cover secondary infection)
- Supportive oxygen; mechanical ventilation generally NOT recommended (poor outcomes)
- ICU admission often futile — goals-of-care discussion essential
- Prognosis: In-hospital mortality ~50%; median survival after AE-IPF ~3–4 months
Monitoring
Patients should be evaluated every 3–6 months (more frequently if progressing) with:
- Spirometry + DLCO
- 6MWT
- HRCT (annually or if significant change)
- Assessment for transplant candidacy
Markers of disease progression / poor prognosis:
- FVC decline ≥10% over 6–12 months
- DLCO decline ≥15%
- Desaturation during 6MWT
- Hospitalization for respiratory cause
- Development of pulmonary hypertension
Prognosis
| Parameter | Data |
|---|---|
| Median survival from diagnosis | 3–5 years |
| 5-year mortality | ~80% |
| Cause of death | Respiratory failure (most common), AE-IPF, lung cancer, cardiovascular |
IPF follows a variable course — most patients show slow, steady decline; some have accelerated decline with AEs; a minority are stable for years.
Key Guidelines
- ATS/ERS/JRS/ALAT 2022 Clinical Practice Guidelines — the most current evidence-based guideline (ATS/ERS, p. 2, 18)
- Available at: American Journal of Respiratory and Critical Care Medicine
Summary Algorithm
Suspected IPF (older adult, bilateral basal crackles, dyspnea)
↓
Exclude alternative ILD causes (CTD serology, drug Hx, exposure Hx)
↓
HRCT
├── Typical UIP → Diagnose IPF (no biopsy needed)
├── Probable UIP → Consider biopsy / genomic classifier / MDD
└── Indeterminate → Biopsy (TBLC or surgical) → MDD
↓
Confirm IPF
↓
Start antifibrotic (nintedanib or pirfenidone)
+ O₂, pulmonary rehab, GERD treatment
+ Early lung transplant referral if high-risk
+ Monitor every 3–6 months
+ Palliative care integration
This is a shared conversation. Sign in to Orris to start your own chat.