Pathology
Asking for Preferences
Asking for Preferences
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I have all the content I need from Robbins & Kumar. Now let me compile a comprehensive, well-structured answer.
Neoplasia: A Detailed Explanation
Source: Robbins & Kumar Basic Pathology
1. What Is a Neoplasm?
Neoplasia literally means "new growth." A neoplastic cell is one that has been transformed - it replicates incessantly because it has become resistant to the regulatory influences that govern normal cells. All neoplasms depend on the host for nutrition and blood supply, but they grow in an uncoordinated way, independently of the signals that drive normal tissue turnover.
In clinical usage, a neoplasm is called a tumor, and the study of tumors is oncology. The single most important distinction is whether a tumor is benign or malignant.
2. Nomenclature
Benign Tumors
Named by attaching the suffix -oma to the cell of origin:
| Cell/Tissue of Origin | Benign Tumor Name |
|---|---|
| Fibrous tissue | Fibroma |
| Cartilage | Chondroma |
| Bone | Osteoma |
| Glandular epithelium | Adenoma |
| Squamous epithelium | Squamous papilloma |
| Blood vessels | Hemangioma |
| Smooth muscle | Leiomyoma |
| Striated muscle | Rhabdomyoma |
Malignant Tumors
| Cell/Tissue of Origin | Malignant Tumor Name |
|---|---|
| Epithelial origin (any) | Carcinoma |
| - Glandular epithelium | Adenocarcinoma |
| - Squamous epithelium | Squamous cell carcinoma |
| Mesenchymal / connective tissue | Sarcoma (e.g., fibrosarcoma, osteosarcoma, leiomyosarcoma) |
| Lymphoid tissue | Lymphoma |
| Plasma cells | Myeloma |
| Embryonal / mixed | Teratoma, blastoma |
Eponymous exceptions (names don't follow the rule): Melanoma (malignant), Hepatoma (malignant hepatocellular carcinoma), Seminoma (malignant).
3. Benign vs. Malignant: Key Distinguishing Features
A. Differentiation and Anaplasia
Differentiation refers to the extent to which tumor cells resemble the normal cells of origin - both morphologically and functionally.
- Benign tumors are well-differentiated. Their cells closely resemble normal counterparts. Example: a lipoma has mature fat cells indistinguishable from normal adipocytes.
- Malignant tumors range from well-differentiated to completely undifferentiated (anaplastic).
Anaplasia (literally "without form") is the hallmark of malignancy. Anaplastic cells show:
- Pleomorphism - variation in cell size and shape
- Hyperchromatic nuclei - dark-staining due to increased DNA content
- High nuclear:cytoplasmic ratio - often 1:1 instead of normal 1:4 to 1:6
- Prominent nucleoli
- Abnormal mitoses - tripolar or multipolar spindles (see image below)
- Tumor giant cells
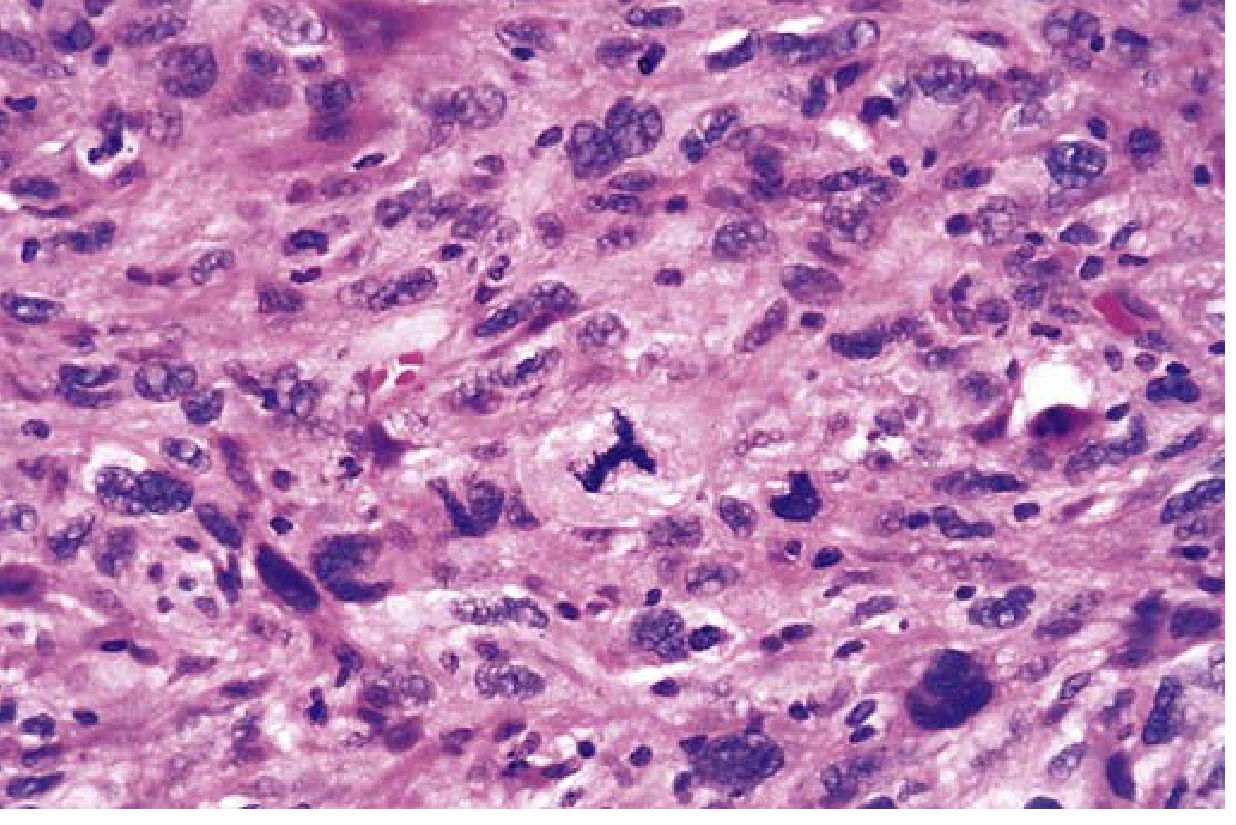
High-power view of anaplastic tumor cells showing pleomorphism and an abnormal tripolar mitotic spindle - Robbins & Kumar Basic Pathology
Dysplasia is disorderly proliferation short of full malignancy. It is characterized by loss of uniformity, architectural disorganization, and nuclear abnormalities. When dysplasia involves the full thickness of an epithelium, it is called carcinoma in situ - a preinvasive stage. Mild-to-moderate dysplasia may regress if the inciting cause is removed.
B. Local Invasion
This is one of the most reliable distinguishing features:
-
Benign tumors grow as cohesive, expansile masses. They develop a rim of compressed fibrous tissue called a capsule, which creates a clear tissue plane - making the tumor discrete, movable, and surgically excisable by enucleation.
- Exception: some benign tumors (hemangiomas, uterine leiomyomas) are not truly encapsulated but are still clearly demarcated.
-
Malignant tumors show progressive infiltration, invasion, and destruction of surrounding tissues. They lack well-defined capsules. Their invasive fingers of tumor cells make complete resection difficult.
C. Metastasis
Metastasis = spread of tumor to a site discontinuous with the primary tumor. It is the feature that most definitively identifies a neoplasm as malignant. Benign tumors never metastasize.
The three main routes of metastasis are:
- Lymphatic spread - most common for carcinomas; to regional lymph nodes first
- Hematogenous spread - most common for sarcomas; tumor cells enter blood vessels; lungs and liver are common sites
- Seeding of body cavities - tumor cells shed into peritoneal, pleural, or pericardial spaces (e.g., ovarian carcinoma seeding the peritoneum)
4. Summary Comparison Table
| Feature | Benign | Malignant |
|---|---|---|
| Differentiation | Well-differentiated | Variable; may be anaplastic |
| Growth rate | Usually slow | Often rapid |
| Mitoses | Rare, normal | Frequent, abnormal |
| Local invasion | Non-invasive; encapsulated | Invasive; no capsule |
| Metastasis | Absent | Present (hallmark) |
| Effect on host | Usually local | Systemic; often fatal |
| Recurrence after surgery | Rare | Common |
5. Carcinogenesis: A Multistep Process
Cancer is fundamentally a genetic disease. Mutations accumulate in somatic cells (or may be inherited), altering genes that regulate growth, survival, and DNA repair. Key points:
- Individual mutations confer selective growth advantages (Darwinian selection)
- Each tumor begins as a single clone - clonal origin
- Over time, tumor progression occurs as further mutations generate more aggressive subclones
The Hallmarks of Cancer (Hanahan & Weinberg)
These are the acquired capabilities that cancer cells develop:
| Hallmark | Mechanism |
|---|---|
| Self-sufficiency in growth signals | Oncogene mutations (RAS, MYC, EGFR); autocrine growth factor loops |
| Insensitivity to growth inhibitors | Loss of tumor suppressors (RB, TP53) |
| Evasion of apoptosis | BCL2 overexpression; TP53 loss |
| Limitless replicative potential | Telomerase reactivation |
| Sustained angiogenesis | VEGF upregulation |
| Invasion and metastasis | Loss of E-cadherin; MMP upregulation; EMT |
| Altered cellular metabolism | Warburg effect (aerobic glycolysis) |
| Evasion of immune surveillance | PD-L1 expression; loss of MHC class I |
6. Key Molecular Players
Proto-Oncogenes → Oncogenes
Proto-oncogenes are normal genes that promote cell growth. When mutated or overexpressed, they become oncogenes that drive uncontrolled proliferation. Oncogenes act as dominant gain-of-function mutations - one mutant allele is sufficient.
Examples:
- RAS - most commonly mutated oncogene in human cancers; point mutations lock it in the "on" position
- MYC - transcription factor; amplified in Burkitt lymphoma, neuroblastoma
- HER2/ERBB2 - receptor tyrosine kinase; amplified in breast cancer
- BCR-ABL - chromosomal translocation in CML; constitutively active tyrosine kinase
Tumor Suppressor Genes
These normally brake cell proliferation. Loss of function (requires both alleles to be inactivated - "two-hit hypothesis") removes the brake.
-
RB (Retinoblastoma protein) - "Governor of the cell cycle." In its active (hypophosphorylated) state, RB binds E2F transcription factors and prevents S-phase entry. Cyclin D/CDK4-6 phosphorylate RB → releases E2F → cell cycle proceeds. In cancer, RB is lost or CDK4 is amplified.
-
TP53 ("Guardian of the Genome") - Most commonly mutated gene in human cancers (>50% of all cancers). Activated by DNA damage:
- Causes G1 arrest (via p21/CDKN1A)
- Induces DNA repair
- If repair fails → triggers apoptosis or senescence
- With TP53 loss, damaged DNA goes unrepaired and mutations accumulate
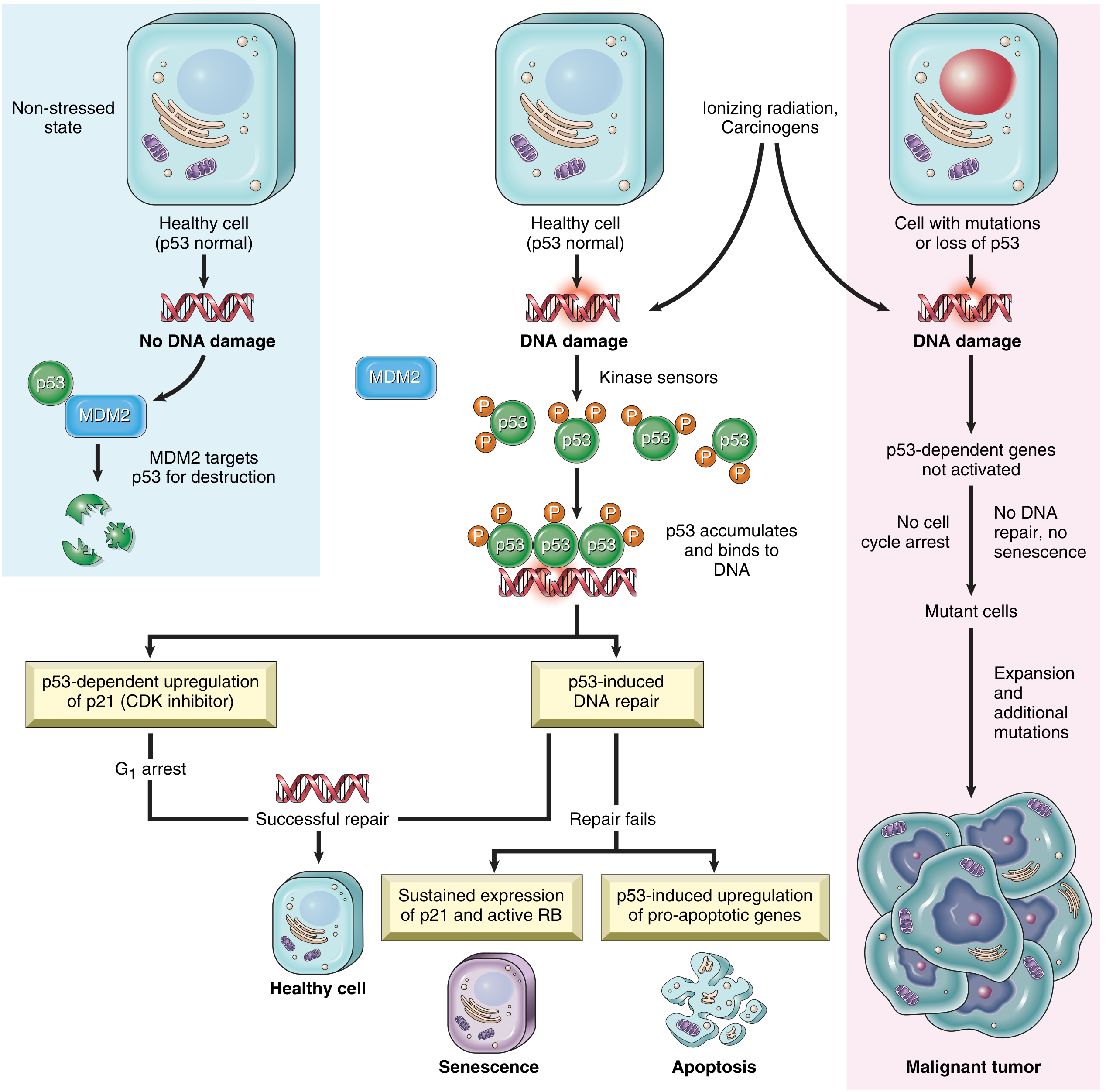
Fig. 6.20 - The role of p53 in maintaining the integrity of the genome. When p53 is lost, DNA damage goes unrepaired and cells progress to malignancy. - Robbins & Kumar Basic Pathology
7. Carcinogens
Chemical Carcinogens
Most are procarcinogens that require metabolic activation to become ultimate carcinogens. They are primarily initiators that cause DNA mutations. Key occupational carcinogens include:
| Agent | Cancer |
|---|---|
| Asbestos | Lung carcinoma, mesothelioma |
| Arsenic | Lung and skin carcinoma |
| Benzene | Acute myeloid leukemia |
| Vinyl chloride | Hepatic angiosarcoma |
| Radon | Lung carcinoma |
Radiation
- Ionizing radiation (UV, X-rays, gamma rays) - causes DNA strand breaks and pyrimidine dimers; directly linked to skin cancers (UV), leukemia (gamma), and thyroid cancer
- UV radiation specifically causes C→T transitions at dipyrimidine sites via thymine dimer formation
Oncogenic Viruses
- HPV (types 16, 18) - cervical, oropharyngeal carcinomas; E6 degrades p53, E7 inactivates RB
- HBV/HCV - hepatocellular carcinoma (chronic inflammation + direct oncogenic effects)
- EBV - Burkitt lymphoma, Hodgkin lymphoma, nasopharyngeal carcinoma
- HTLV-1 - adult T-cell leukemia/lymphoma
- HHV-8 (KSHV) - Kaposi sarcoma
8. Genetic Mechanisms of Cancer
| Mechanism | Example |
|---|---|
| Point mutations | RAS (codon 12/13) - most common in colorectal, pancreatic, lung cancers |
| Gene amplification | HER2 in breast cancer; MYCN in neuroblastoma |
| Chromosomal rearrangement/translocation | BCR-ABL t(9;22) in CML; MYC-IgH t(8;14) in Burkitt lymphoma |
| Deletions | RB1 loss in retinoblastoma; TP53 loss on 17p |
| Epigenetic silencing | Promoter hypermethylation of tumor suppressor genes |
| MicroRNA dysregulation | miR-21 overexpression silences tumor suppressors |
Quick Exam Summary
| Concept | Key Point |
|---|---|
| Neoplasm | Autonomous, purposeless, excessive cell growth |
| Benign | Localized, encapsulated, well-differentiated, no metastasis |
| Malignant | Invasive, metastatic, variable differentiation |
| Carcinoma | Malignant tumor of epithelial origin |
| Sarcoma | Malignant tumor of mesenchymal origin |
| Anaplasia | Hallmark of malignancy - loss of differentiation |
| Metastasis | Definitive proof of malignancy |
| Oncogene | Dominant gain-of-function; accelerates growth (RAS, MYC, HER2) |
| Tumor suppressor | Recessive loss-of-function; brakes growth (RB, TP53) |
| Two-hit hypothesis | Both alleles of TSG must be lost (Knudson) |
| Most mutated gene | TP53 (>50% of all cancers) |
| Warburg effect | Aerobic glycolysis - glucose → lactate even in O2 |
- Robbins & Kumar Basic Pathology, Chapter 6 (Neoplasia), pp. 214-236
This is a shared conversation. Sign in to Orris to start your own chat.