Pathogenesis of diabetic kidney disease
Reading File
Finding Sources
Searching PubMed
"diabetic kidney disease" AND pathogenesis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Searching the Web
diabetic kidney disease pathogenesis mechanisms 2024 2025 review
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have rich content from multiple authoritative sources. Let me compile the comprehensive answer.
Pathogenesis of Diabetic Kidney Disease (DKD)
Diabetic kidney disease (DKD), also called diabetic nephropathy, is the leading cause of end-stage kidney disease (ESKD) globally, affecting 30-40% of patients with both type 1 and type 2 diabetes. The pathogenesis is multifactorial, involving metabolic, hemodynamic, inflammatory, genetic, and epigenetic mechanisms that converge to produce progressive glomerular and tubulointerstitial injury.
1. Central Initiating Trigger: Chronic Hyperglycemia
Hyperglycemia is the necessary prerequisite. As Harrison's states: "Like other microvascular complications, the pathogenesis of diabetic nephropathy is related to chronic hyperglycemia." Hyperglycemia activates multiple downstream injury pathways - Brenner and Rector's The Kidney, p. 1588.
2. Metabolic Pathways Activated by Hyperglycemia
A. Polyol Pathway
Excess glucose is shunted through aldose reductase into sorbitol, consuming NADPH (reducing oxidative defense) and ultimately depleting intracellular myoinositol. This alters Na+/K+-ATPase activity and promotes cellular injury.
B. Protein Kinase C (PKC) Activation
Hyperglycemia increases intracellular diacylglycerol (DAG), activating PKC isoforms (particularly PKC-β). PKC activation leads to:
- Increased VEGF expression (enhancing vascular permeability)
- Upregulation of TGF-β (promoting fibrosis)
- Activation of NAD(P)H oxidase (generating reactive oxygen species)
- Reduced eNOS activity (impairing vasodilation)
C. Advanced Glycation End Products (AGEs) and RAGE
Non-enzymatic glycation of proteins and lipids forms AGEs. AGEs bind to their receptor (RAGE), activating NF-κB and downstream proinflammatory and profibrotic cascades. AGEs directly crosslink GBM proteins and alter mesangial cell function. Endothelin-1, stimulated by the AGE-RAGE axis, contributes to oxidative stress in the diabetic kidney - Brenner and Rector's The Kidney (Endothelin system chapter), p. 2738.
D. Hexosamine Pathway
Glucose flux through the hexosamine pathway modifies transcription factors via O-linked N-acetylglucosamine (O-GlcNAc), altering expression of TGF-β, PAI-1, and other profibrotic genes.
E. Reactive Oxygen Species (ROS) and Oxidative Stress
Mitochondrial overproduction of superoxide (from glucose oxidation) is considered a "unifying mechanism" activating all four above pathways. ROS cause direct cellular damage and activate NFκB. Recent evidence shows hyperglycemia disrupts mitochondrial dynamics - upregulating fission proteins (Drp1) and suppressing fusion proteins (Mfn2), leading to mitochondrial fragmentation and impaired function, as well as compromised mitophagy through PINK1/Parkin and BNIP3/Nix pathways.
3. Hemodynamic Alterations
Glomerular Hyperfiltration
In the early phase, hyperglycemia and glucosuria activate tubuloglomerular feedback: glucose-mediated tubular Na+ reabsorption reduces macula densa NaCl, causing afferent arteriolar dilation and increased GFR. This glomerular hyperfiltration is a hallmark of early DKD and mediates mechanical stretch injury to the glomerular capillary wall.
RAAS Activation
Angiotensin II (Ang II) plays a central role:
- Preferentially constricts the efferent arteriole, raising intraglomerular pressure
- Directly stimulates TGF-β and aldosterone
- Promotes mesangial cell contraction and matrix production
- Activates NADPH oxidase, generating ROS
ACE gene D allele (high enzymatic activity) is associated with greater susceptibility to ESKD, and RAAS blockers remain standard therapy - Brenner and Rector's The Kidney, p. 3381.
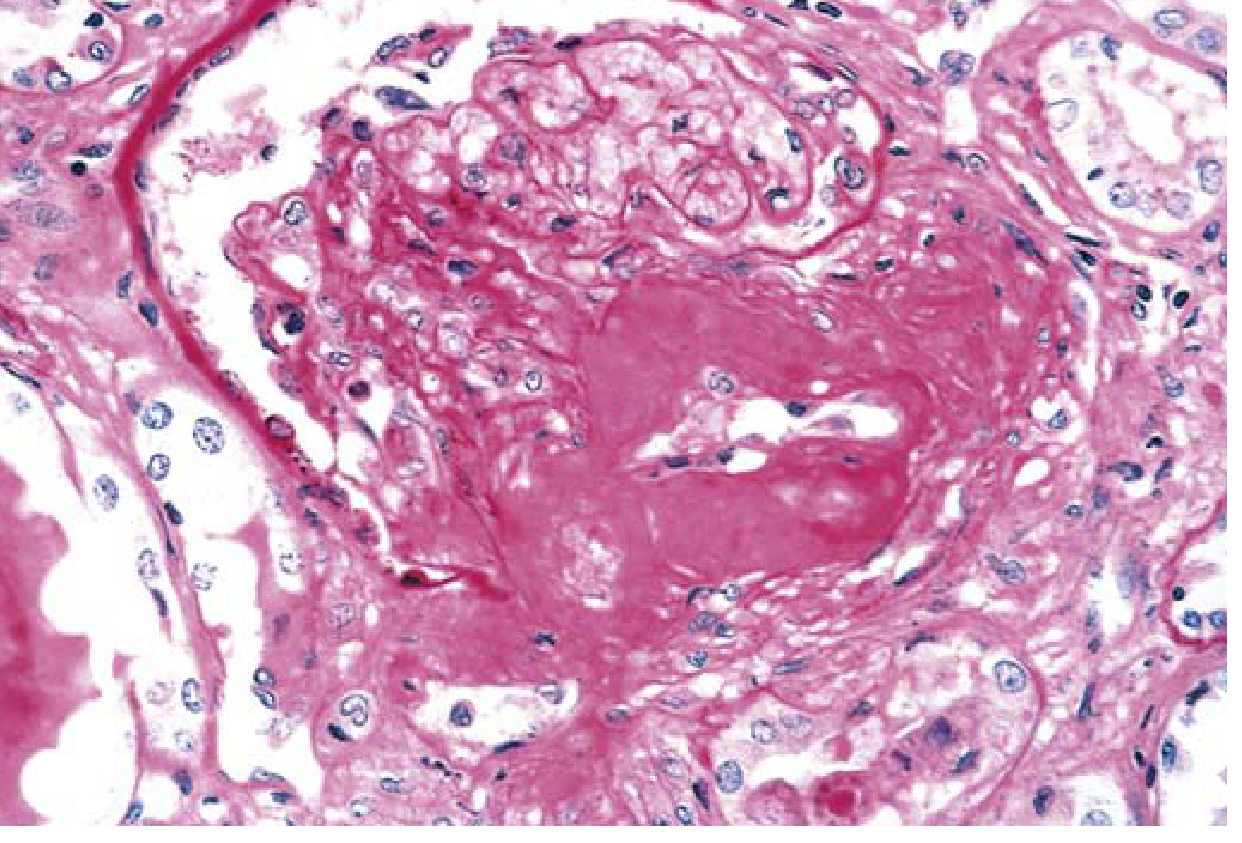
Hyaline Arteriolosclerosis
DKD uniquely affects both the afferent and efferent arterioles (efferent involvement is virtually pathognomonic of diabetes). This vascular compromise contributes to ischemia and nephrosclerosis - Robbins & Kumar Basic Pathology, p. 751.
4. Structural Lesions (Glomerulopathy)
Glomerular Basement Membrane (GBM) Thickening
The earliest ultrastructural change, detectable by EM within a few years of diabetes onset, even before any functional change. GBM thickness correlates with diabetes duration and progressive albuminuria.
Mesangial Expansion
Increased mesangial matrix deposition + mesangial cell proliferation. Found in the majority after >10 years of diabetes. When severe, produces nephrotic syndrome.
Nodular Glomerulosclerosis (Kimmelstiel-Wilson Lesion)
Ball-like deposits of laminated matrix in the periphery of the glomerulus. Present in approximately 15-30% of patients with long-term diabetes. The nodule forms via mesangiolysis - fraying of the mesangium leads to microaneurysm formation and matrix accumulation around it. This lesion is virtually pathognomonic of diabetes - Robbins & Kumar Basic Pathology, p. 751.
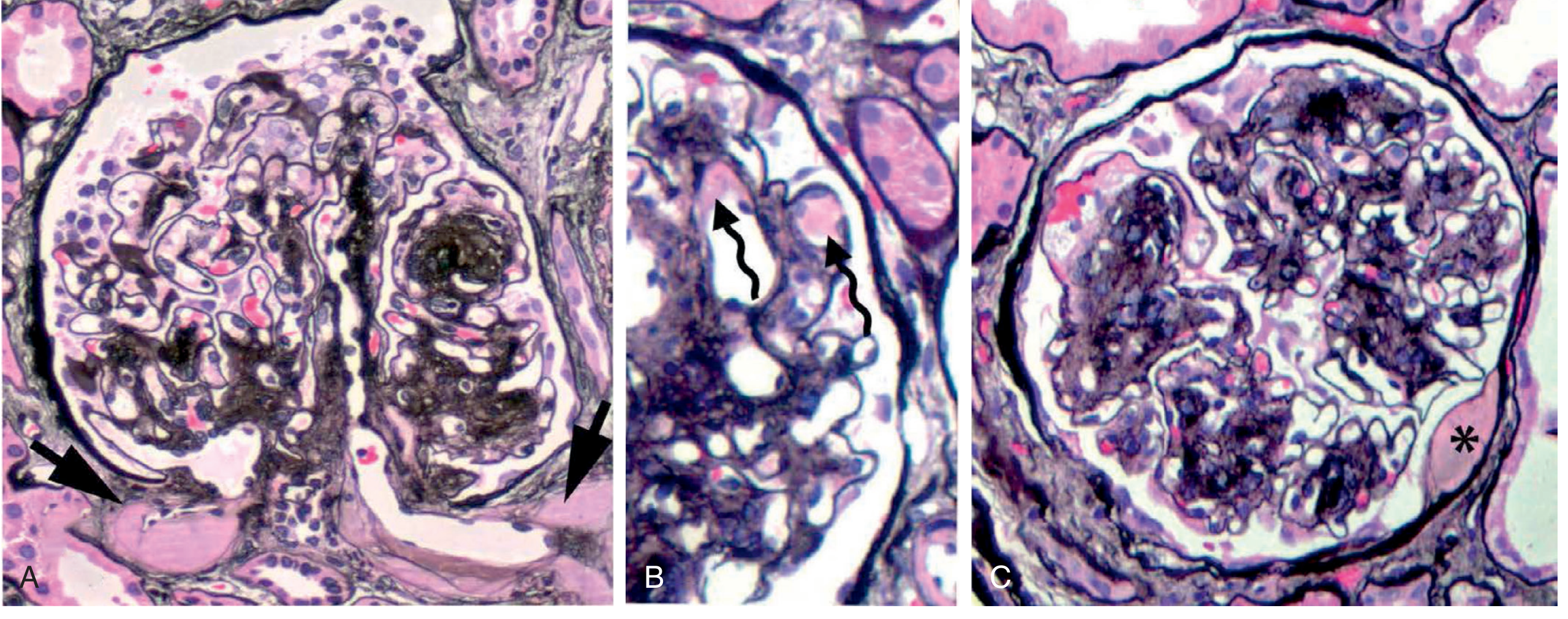
Exudative lesions: (A) hyalinosis of both afferent and efferent arterioles, (B) fibrin caps in capillaries, and (C) capsular drop - Brenner and Rector's The Kidney
5. Podocyte Injury and Loss
Podocytes are terminally differentiated and cannot regenerate. In DKD:
- Foot process effacement (widening) is detectable even in normoalbuminuric patients
- Increased urine nephrin excretion signals early podocyte injury before microalbuminuria
- Podocyte detachment from the GBM occurs early and worsens with albuminuria
- Mechanisms of podocyte loss: apoptosis (glucose-induced oxidative stress), autophagy dysregulation, reduced αβ1-integrin expression, and detachment
- Podocyte loss leads to secondary FSGS, with a predilection for the glomerulotubular junction - Brenner and Rector's The Kidney, p. 1779
Podocyte number per glomerulus in microalbuminuric Pima Indians was the strongest single predictor of progression to overt nephropathy.
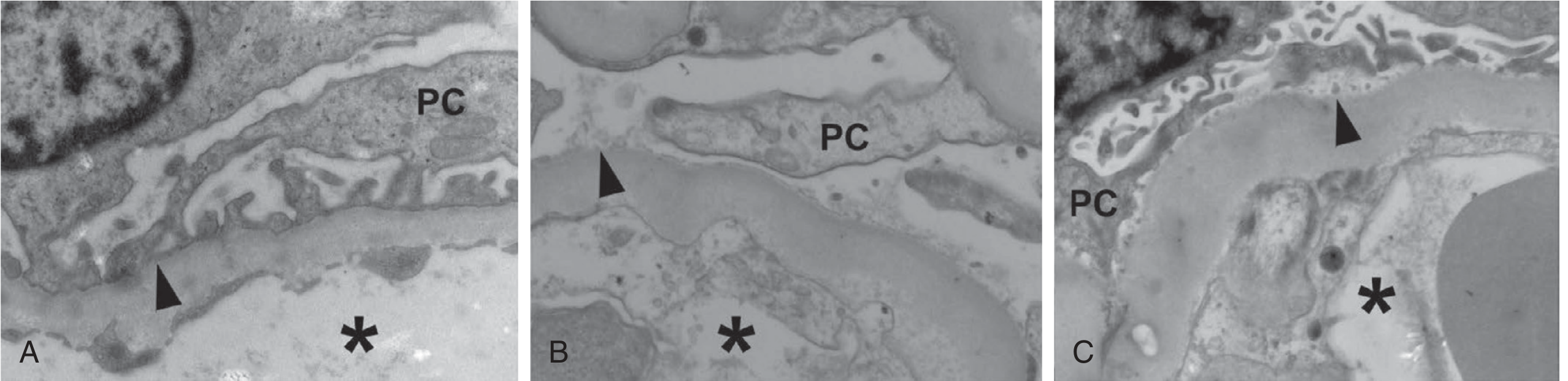
6. Tubular Injury and Tubulointerstitial Fibrosis
The proximal tubule is the major site of early tubular injury in DKD:
- Glucose-induced proximal tubular hypertrophy and hyperfiltration
- Tubular exposure to filtered proteins (proteinuria-induced tubulotoxicity) activates NF-κB and TGF-β, driving interstitial inflammation and fibrosis
- Epithelial-to-mesenchymal transition (EMT) contributes to myofibroblast accumulation
- Biomarkers: urine KIM-1, NGAL, L-FABP, NAG reflect tubular damage early
7. Inflammatory Mechanisms
DKD is now recognized as an inflammatory disease:
- Monocyte/macrophage infiltration in glomeruli and interstitium
- IL-6, IL-10 gene polymorphisms influence susceptibility
- ICAM-1 (intercellular adhesion molecule-1) polymorphisms increase risk - Brenner and Rector's The Kidney, p. 3382
- NF-κB activation drives cytokine and chemokine release
- Recent evidence highlights ferroptosis (iron-mediated lipid peroxidation) activating TGF-β/Smad3 and NF-κB pathways, creating a feed-forward fibrotic loop
8. Growth Factors and Cytokines
| Factor | Role in DKD |
|---|---|
| TGF-β | Master profibrotic cytokine; promotes ECM synthesis by mesangial and tubular cells |
| VEGF | Increases glomerular permeability; PKC-mediated |
| Angiotensin II | Efferent arteriolar constriction, TGF-β upregulation, ROS generation |
| Endothelin-1 | Contributes to oxidative stress, vasoconstriction |
| CTGF | Downstream of TGF-β; promotes fibrosis |
| IL-6, TNF-α | Inflammatory amplification |
9. Dyslipidemia as a Co-Pathogen
- Oxidized LDL and free fatty acids cause direct podocyte mitochondrial dysfunction and ROS accumulation
- Intraglomerular lipid droplets (cholesterol + triglycerides) activate pro-sclerotic, proliferative, and proinflammatory cytokines
- ABCA1 (cholesterol efflux transporter) is downregulated in DKD glomeruli, correlating with reduced eGFR and podocyte lipid accumulation - Brenner and Rector's The Kidney, p. 1769
10. Epigenetic Mechanisms and Metabolic Memory
Even after normalization of blood glucose (e.g., after pancreas transplantation), DKD can continue to progress - the phenomenon of metabolic memory. Mechanisms include:
- DNA cytosine methylation changes in fibrosis-related genes
- Histone post-translational modifications (H3K4me1, H3K9me2)
- Non-coding RNAs: miRNAs (miR-21, miR-192, miR-93) and long non-coding RNAs modulate TGF-β signaling and fibrotic gene expression
This cellular memory explains the long-lasting benefits of early glycemic control observed in DCCT (type 1) and UKPDS (type 2) trials - Brenner and Rector's The Kidney, p. 1772.
11. Natural History / Staging
The classical course (initially defined for type 1 DM but applicable to type 2):
| Stage | GFR | Albuminuria | Structural Changes |
|---|---|---|---|
| Hyperfiltration/hypertrophy (0-2 yrs) | Elevated | Normal | Renal enlargement |
| Silent (2-5 yrs) | Returns to normal | Normal | GBM thickening, mesangial expansion |
| Microalbuminuria (5-15 yrs) | Normal or slightly ↓ | 30-299 mg/g (ACR) | Progressive glomerulopathy |
| Overt proteinuria / macroalbuminuria | Declining | ≥300 mg/g | Nodular glomerulosclerosis |
| ESKD | <15 mL/min | Heavy proteinuria | Global glomerulosclerosis, tubulointerstitial fibrosis |
(Harrison's Principles, p. 1592)
12. Genetic Susceptibility
- Only ~30-40% of diabetics develop DKD, indicating genetic predisposition
- ACE gene insertion/deletion (I/D) polymorphism: DD genotype associated with higher ACE activity and ESKD susceptibility
- ACACB gene (acetyl-CoA carboxylase β) polymorphisms linked to proinflammatory tubular responses
- IL-10, IL-6 gene polymorphisms affect susceptibility
- GWAS studies continue to identify additional loci
Histological Image: Severe Hyaline Arteriolosclerosis
Summary of Key Pathways
Hyperglycemia
|
├── Polyol pathway → sorbitol accumulation, NADPH depletion
├── PKC activation → VEGF↑, TGF-β↑, ROS↑
├── AGE-RAGE axis → NF-κB activation, GBM crosslinking
├── Hexosamine pathway → profibrotic gene expression
└── Mitochondrial ROS (unifying mechanism)
|
┌─────────┴──────────┐
Hemodynamic changes Structural changes
| |
RAAS activation GBM thickening
Glomerular hyperfiltration Mesangial expansion
Intraglomerular HTN Kimmelstiel-Wilson nodules
| Podocyte loss → FSGS
└─────────┬──────────┘
|
Inflammation & fibrosis
(TGF-β, macrophage infiltration, ferroptosis)
|
Tubulointerstitial fibrosis
|
ESKD
Key Sources:
- Brenner and Rector's The Kidney (Chapter 39) - the most detailed nephrology reference
- Robbins & Kumar Basic Pathology, p. 751
- Harrison's Principles of Internal Medicine 22E, p. 1590-1592
- Recent molecular reviews: Frontiers in Pharmacology 2025 and PMC molecular pathways review
This is a shared conversation. Sign in to Orris to start your own chat.