Type of sputum
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Here is a complete, textbook-sourced breakdown of the types of sputum and their clinical significance:
Types of Sputum
1. Mucoid
- Appearance: Clear, white, or grey; viscid and non-offensive
- Significance: Produced in excess mucus states without active infection
- Associated conditions: Chronic bronchitis (stable phase), asthma, bronchiectasis (between exacerbations)
2. Purulent
- Appearance: Yellow or green, thick; due to neutrophils and cellular debris
- Significance: Reliable indicator of infection in the tracheobronchial tree or lungs
- Associated conditions: Acute bacterial pneumonia, bronchiectasis exacerbation, lung abscess
3. Mucopurulent
- Appearance: Mixed - partially mucoid, partially purulent
- Significance: Transition between stable and infected states
- Associated conditions: Acute exacerbation of chronic bronchitis (COPD exacerbation - Haemophilus influenzae, Moraxella catarrhalis, Streptococcus pneumoniae are common pathogens), bronchiectasis
4. Rust-Colored (Rusty)
- Appearance: Brown-orange ("rusty"); caused by even dispersion of blood in yellow, purulent sputum - as stated in Fishman's Pulmonary Diseases and Disorders
- Significance: Classic sign of lobar consolidation
- Associated conditions: Pneumococcal pneumonia (the hallmark) - "The classic presentation of pneumococcal pneumonia consists of... production of rusty or mucoid sputum" - Murray & Nadel's Textbook of Respiratory Medicine
5. Currant Jelly
- Appearance: Red-black, thick, gelatinous, blood-mixed
- Significance: Necrotic lung tissue mixed with blood
- Associated conditions: Klebsiella pneumoniae pneumonia - "The classic description of sputum in Klebsiella pneumonia is a resemblance to currant jelly; it also contains blood, but it is bright red and more translucent and viscid than the sputum of pneumococcal pneumonia" - Fishman's Pulmonary Diseases and Disorders
6. Foul-Smelling (Fetid) Purulent
- Appearance: Purulent, foul odor; may form layers on standing
- Significance: Anaerobic bacterial infection
- Associated conditions: Lung abscess, bronchiectasis (severe), anaerobic pneumonia (commonly streptococci) - "Purulent sputum with a foul odor usually indicates an anaerobic infection" - Fishman's Pulmonary Diseases and Disorders
7. Pink Frothy
- Appearance: Blood-tinged foam, pink and frothy
- Significance: Alveolar flooding with blood-stained transudate
- Associated conditions: Acute pulmonary edema (left ventricular failure, mitral valve disease, aspiration chemical pneumonitis, negative-pressure pulmonary edema) - well-documented across multiple textbooks (Fishman's, Fuster & Hurst's The Heart, Barash's Clinical Anesthesia)
8. Blood-Stained / Hemoptysis
- Appearance: Frankly blood-containing, ranging from streaks to massive
- Significance: Mucosal or vascular disruption in the airways/lungs
- Associated conditions:
- Streaky blood - bronchitis, bronchogenic carcinoma (recurrent small hemoptysis common)
- Massive hemoptysis - TB, bronchiectasis, pulmonary infarction, lung abscess
9. Serous / Watery
- Appearance: Thin, watery, frothy but not pink
- Significance: Fluid transudation
- Associated conditions: Adenocarcinoma in situ (formerly BAC) - "large quantities of watery, mucoid sputum" - Fishman's; early pulmonary edema
10. Hemosiderin-Laden ("Heart Failure" Sputum)
- Appearance: Brown-flecked; contains hemosiderin-laden macrophages
- Significance: Chronic pulmonary venous congestion
- Associated conditions: Chronic left heart failure, mitral stenosis
11. Black / Dark
- Appearance: Black or dark grey
- Significance: Inhaled carbonaceous material
- Associated conditions: Coal workers' pneumoconiosis (anthracosis), heavy smokers
12. Anchovy Sauce
- Appearance: Thick, dark-brown, resembling anchovy sauce
- Significance: Hepatic abscess contents entering the lung
- Associated conditions: Hepatic amoebiasis with pleuropulmonary extension (amoebic empyema/lung abscess)
Quick Reference Table
| Sputum Type | Color/Appearance | Key Condition |
|---|---|---|
| Mucoid | Clear/white | Chronic bronchitis, asthma |
| Purulent | Yellow/green | Bacterial pneumonia, abscess |
| Mucopurulent | Mixed | COPD exacerbation, bronchiectasis |
| Rusty | Brown-orange | Pneumococcal pneumonia |
| Currant jelly | Red-black, viscid | Klebsiella pneumonia |
| Foul purulent | Foul-smelling | Anaerobic infection, lung abscess |
| Pink frothy | Pink foam | Pulmonary edema |
| Hemoptysis | Frank blood | TB, malignancy, PE |
| Watery/serous | Clear, watery | Adenocarcinoma in situ |
| Hemosiderin | Brown-flecked | Chronic LVF, mitral stenosis |
| Black | Black/dark | Pneumoconiosis |
| Anchovy sauce | Dark brown | Amoebic lung abscess |
Sources: Fishman's Pulmonary Diseases and Disorders; Murray & Nadel's Textbook of Respiratory Medicine; Fuster & Hurst's The Heart, 15th Ed; Barash's Clinical Anesthesia, 9th Ed.
Bronchitis and bronchiectasis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a comprehensive, textbook-sourced breakdown of both conditions:
Bronchitis
A. Chronic Bronchitis
Definition
Chronic bronchitis is defined clinically as a persistent productive cough for at least 3 consecutive months in at least 2 consecutive years, in the absence of any other identifiable cause. - Robbins, Cotran & Kumar Pathologic Basis of Disease
Etiology / Pathogenesis
The primary initiating factor is exposure to inhaled irritants. Three mechanisms are involved:
- Mucus hypersecretion - the earliest feature; caused by enlargement of submucosal mucus-secreting glands in the trachea and bronchi, driven by mediators including histamine and IL-13. Goblet cells increase in small airways as well.
- Acquired CFTR dysfunction - cigarette smoke leads to acquired CFTR dysfunction, causing secretion of abnormal, dehydrated mucus that worsens chronic bronchitis.
- Inflammation - inhaled irritants cause neutrophil, lymphocyte, and macrophage infiltration. Unlike asthma, eosinophils are NOT seen.
- Infection - does not initiate chronic bronchitis but maintains it and triggers acute exacerbations.
Key causes:
- Cigarette smoking (90% of cases)
- Air pollutants (sulfur dioxide, nitrogen dioxide)
- Grain dust, cotton dust, silica
Morphology
Gross:
- Hyperemic, swollen, edematous mucosal lining of large airways
- Mucinous or mucopurulent secretions covering the lumen
- Smaller bronchi and bronchioles filled with secretions
Microscopic:
- Enlargement of mucus-secreting submucosal glands - assessed by the Reid Index (ratio of gland layer thickness to bronchial wall thickness between epithelium and cartilage). Normal = 0.4; elevated in chronic bronchitis, proportional to severity.
- Goblet cell hyperplasia in small airways
- Smooth muscle hypertrophy, peribronchial fibrosis
- Chronic inflammatory cells (lymphocytes, macrophages)
- Mucus plugging, inflammation, and fibrosis leading to bronchiolar narrowing
- In severe cases: bronchiolitis obliterans (complete obliteration of bronchiolar lumen by fibrosis)
Clinical Features
- Persistent productive cough with mucoid sputum (early); becomes mucopurulent during exacerbations
- Progressive airflow obstruction (small airway disease)
- May develop chronic outflow obstruction with associated emphysema
- Advanced disease: hypoxemia, pulmonary hypertension, cor pulmonale
- Common exacerbation pathogens: Haemophilus influenzae, Moraxella catarrhalis, Streptococcus pneumoniae
B. Acute Bronchitis
- Acute self-limiting inflammation of the trachea and bronchi
- Mostly viral (rhinovirus, influenza, parainfluenza, RSV)
- Presents with cough, low-grade fever, mucoid or mucopurulent sputum
- Usually resolves within 2-3 weeks; antibiotics not routinely indicated
Bronchiectasis
Definition
Bronchiectasis is a pathologic and permanent dilation of bronchi with bronchial wall thickening, caused by destruction of smooth muscle and elastic tissue by chronic infection and inflammation. - Schwartz's Principles of Surgery, 11th Ed; Robbins, Cotran & Kumar
Etiology / Causes
Congenital / hereditary:
- Cystic fibrosis - most important; defective CFTR causes thick viscid secretions, impaired mucociliary clearance, chronic bacterial infection, and widespread bronchial wall destruction
- Primary ciliary dyskinesia (Kartagener syndrome) - autosomal recessive; dynein arm mutations impair cilia, preventing clearance; ~50% have situs inversus + bronchiectasis + sinusitis; males are infertile due to sperm dysmotility
- Immunodeficiency states - selective IgA deficiency, hypogammaglobulinemia
- Intralobar pulmonary sequestration
Acquired - infectious:
- Necrotizing bacterial pneumonia (postpneumonia bronchiectasis)
- Pertussis, measles, severe influenza, varicella pneumonia
- Tuberculosis (upper lobe predilection)
Acquired - obstructive:
- Tumors, foreign body, impacted mucus - produce localized bronchiectasis distal to obstruction
Acquired - immunologic/inflammatory:
- Allergic bronchopulmonary aspergillosis (ABPA) - Th2 response to Aspergillus fumigatus, high IgE, eosinophilic airway inflammation, mucus plugs
- Rheumatoid arthritis, SLE, inflammatory bowel disease
- Post-lung transplant (chronic rejection), post-HSCT (graft-vs-host disease)
Up to 50% of cases are idiopathic.
Pathogenesis - "Vicious Cycle"
The key mechanism is a self-perpetuating vicious cycle (Murray & Nadel's):
- Airway epithelial and ciliary dysfunction + mucus hypersecretion
- Chronic infection induces further mucus hypersecretion
- Inflammation causes permanent airway injury and dilation
- Bronchiectatic airways have poor mucociliary clearance → perpetuates infection and inflammation
Common colonizing organisms:
- Haemophilus influenzae (~50-55%)
- Pseudomonas aeruginosa (12-30%)
- Streptococcus pneumoniae (~12%)
- Nontuberculous mycobacteria
Types (Morphologic Classification)
Three principal types based on pathologic morphology (Schwartz's Principles of Surgery):
| Type | Description |
|---|---|
| Cylindrical | Uniformly dilated bronchi; mildest form |
| Varicose | Irregular or beaded pattern of dilation |
| Saccular (Cystic) | Peripheral balloon-type dilation; most severe; most common after obstruction/infection |
Morphology
Gross:
- Lower lobes bilaterally affected in most cases (except TB which affects upper lobes)
- Airways dilated up to 4x normal size; can be followed almost to the pleural surface (normally bronchioles are not visible within 2-3 cm of pleura)
- On cut surface: dilated bronchi appear cystic, filled with mucopurulent secretions
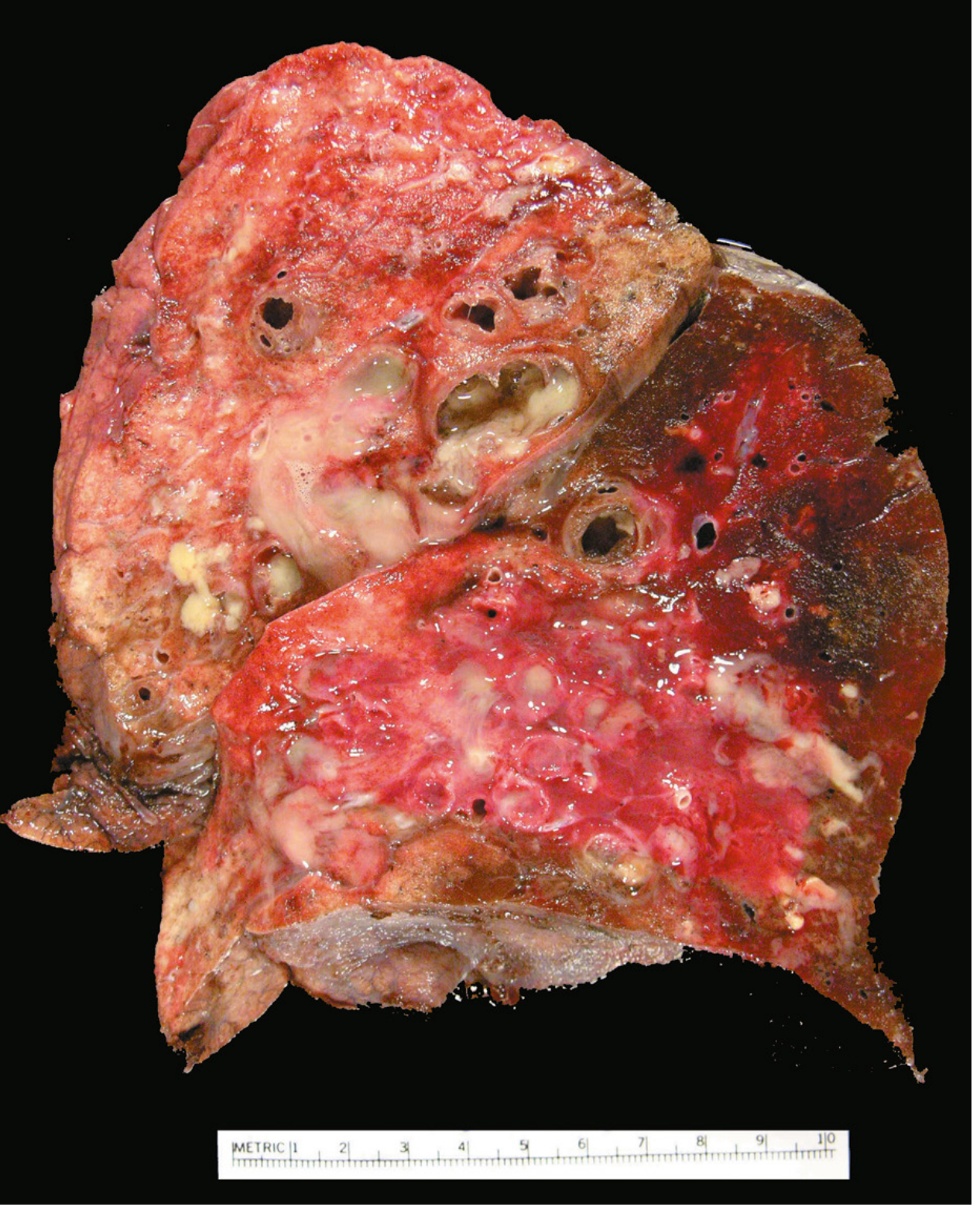
Microscopic:
- Acute and chronic inflammatory exudate within bronchial/bronchiolar walls
- Desquamation of lining epithelium with extensive ulceration
- Squamous metaplasia of remaining epithelium (further impairs clearance)
- Necrosis may form abscess cavities
- Fibrosis of bronchial/bronchiolar walls in chronic cases → bronchiolar obliteration
- In ABPA: fungal hyphae visible on special stains within mucoid plugs
Clinical Features
- Severe persistent cough with copious foul-smelling, sometimes bloody sputum
- Daily sputum volume 10 mL to >150 mL, correlating with disease extent
- Paroxysms of cough on waking - postural drainage of accumulated secretions
- Progressive dyspnea and orthopnea in severe cases
- Hemoptysis (may be massive - erosion of hypertrophied bronchial arteries)
- "Dry bronchiectasis" - nonproductive cough; upper lobe involvement (common in TB)
- Episodes precipitated by upper respiratory infections
Complications (now less frequent with antibiotics):
- Cor pulmonale
- Brain abscesses
- Secondary amyloidosis
Diagnosis
- HRCT chest - modality of choice; shows dilated thick-walled bronchi, "signet ring sign," "train tracks"
- CXR: hyperinflation, bronchiectatic cysts, dilated thick-walled bronchi
- Sputum culture and sensitivity
- Sputum AFB smear/culture (for nontuberculous mycobacteria)
- Spirometry - obstructive pattern
Management (Schwartz's Principles of Surgery)
Medical:
- Airway clearance - chest physiotherapy (percussion, vibration, postural drainage)
- Bronchodilators for reversible airflow limitation
- Antibiotics for exacerbations - 2-3 weeks of broad-spectrum IV antibiotics (tailored to culture), followed by oral course
- Macrolide antibiotics - decrease sputum production, inhibit cytokine release, inhibit neutrophil adhesion, disrupt Pseudomonas biofilm
- Hypertonic saline (7%) - improves mucociliary clearance, reduces exacerbations, improves FEV₁ by 15%
- Inhaled antibiotics (tobramycin, colistin) - improve bacterial clearance
Surgical:
- Resection of localized bronchiectatic segment/lobe for refractory symptoms
- Multifocal disease must be excluded before surgery
- Bronchial artery embolization for massive hemoptysis (preferred over surgery)
- Bilateral lung transplant for end-stage disease
Quick Comparison
| Feature | Chronic Bronchitis | Bronchiectasis |
|---|---|---|
| Definition | Clinical (productive cough ≥3 months/year × 2 years) | Anatomical (permanent bronchial dilation) |
| Key pathology | Mucous gland hypertrophy, Reid index elevated | Bronchial wall destruction, permanent dilation |
| Sputum | Mucoid; mucopurulent in exacerbations | Copious, foul-smelling, purulent; may be bloody |
| Main cause | Cigarette smoking | Infection, congenital (CF, PCD), obstruction |
| Distribution | Large and small airways | Lower lobes bilaterally (except TB) |
| Hallmark sign | Reid index > 0.4 | Signet ring sign / tram-track on HRCT |
| Key complication | COPD, cor pulmonale | Hemoptysis, brain abscess, amyloidosis |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Schwartz's Principles of Surgery, 11th Ed; Murray & Nadel's Textbook of Respiratory Medicine; Fishman's Pulmonary Diseases and Disorders
Bleeding diathesese
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Excellent content gathered. Now I'll compile the full, organized answer.
Bleeding Diatheses
Bleeding diatheses (hemorrhagic disorders) are conditions characterized by abnormal or excessive bleeding that may be spontaneous or triggered by minor trauma/surgery. They result from defects in one or more components of hemostasis: vessels, platelets, or coagulation factors.
Diagnostic Laboratory Tests (Overview)
Before classifying, understanding the key tests is essential - Robbins & Kumar Basic Pathology:
| Test | What it measures | Prolonged in |
|---|---|---|
| PT (Prothrombin Time) | Extrinsic + common pathway | Factors V, VII, X, prothrombin, fibrinogen deficiency; warfarin |
| PTT (Partial Thromboplastin Time) | Intrinsic + common pathway | Factors V, VIII, IX, X, XI, XII, prothrombin, fibrinogen deficiency; heparin; hemophilia |
| Platelet count | Quantitative platelet assessment | Thrombocytopenia (<150,000/μL) |
| Platelet function tests | Qualitative platelet defects | vWD, uremia, aspirin, myeloproliferative disorders |
| Fibrin split products (D-dimer) | Fibrinolysis activation | DIC |
Classification of Bleeding Diatheses
I. Vascular / Connective Tissue Disorders
Bleeding results from vascular fragility - laboratory coagulation tests are usually normal.
Features: Spontaneous petechiae and ecchymoses in skin and mucous membranes from minor trauma only.
Causes:
- Vitamin C deficiency (Scurvy) - defective collagen synthesis in vessel walls
- Amyloidosis affecting blood vessels
- Chronic glucocorticoid use - connective tissue atrophy
- Rare inherited connective tissue disorders (Ehlers-Danlos syndrome, Marfan syndrome)
- Infectious/hypersensitivity vasculitides - Henoch-Schonlein purpura (IgA vasculitis), meningococcemia (DIC/petechiae from endothelial damage)
- Senile purpura - atrophic connective tissue in elderly
II. Platelet Disorders (Primary Hemostasis Defects)
Clinical pattern: Petechiae, ecchymoses, epistaxis, gum bleeding, menorrhagia, excessive bleeding from minor wounds. Petechiae are characteristic.
A. Thrombocytopenia (Quantitative Defects)
Platelet count < 150,000/μL. Spontaneous bleeding when < 5,000/μL; post-traumatic bleeding risk at 20,000-50,000/μL.
Causes (Table - Robbins & Kumar):
Decreased Production:
- Generalized bone marrow failure: aplastic anemia, marrow infiltration (leukemia, disseminated cancer)
- Selective impairment: drug-induced (alcohol, thiazides, cytotoxic drugs), infections (measles, HIV)
- Ineffective megakaryopoiesis: megaloblastic anemia, paroxysmal nocturnal hemoglobinuria (PNH)
Decreased Survival - Immunologic:
- Immune Thrombocytopenic Purpura (ITP)
- Autoimmune: SLE
- Alloimmune: post-transfusion, neonatal
- Drug-associated: quinidine, heparin (HIT), sulfa compounds
- Infections: EBV, HIV, CMV
Decreased Survival - Non-Immunologic:
- DIC
- TTP (Thrombotic Thrombocytopenic Purpura)
- HUS (Hemolytic Uremic Syndrome)
- Microangiopathic hemolytic anemias
Sequestration:
- Hypersplenism
Dilutional:
- Multiple blood transfusions for massive blood loss
Immune Thrombocytopenic Purpura (ITP) - Key Details
- Chronic ITP: Most common in women 20-40 years. Autoantibodies (IgG) against platelet membrane glycoproteins IIb/IIIa or Ib/IX - detected in ~80% of cases. Spleen is the major site of both antibody production and IgG-coated platelet destruction.
- Bone marrow: Increased megakaryocytes (compensatory).
- Features: Petechiae, easy bruising, epistaxis, gum bleeding, hemorrhage after minor trauma. Serious intracranial hemorrhage is rare.
- Treatment: Glucocorticoids, IV immunoglobulin. Splenectomy (normalizes platelet count in >2/3 of patients).
- Acute ITP (children): Post-viral, self-limited.
Thrombotic Thrombocytopenic Purpura (TTP)
Classic pentad:
- Thrombocytopenia
- Microangiopathic hemolytic anemia (MAHA) - fragmented RBCs (schistocytes)
- Renal failure
- Fever
- CNS involvement
Pathogenesis: Deficiency of ADAMTS13 (a metalloprotease that cleaves unusually large vWF multimers). Without ADAMTS13, hyperactive very-high-molecular-weight vWF multimers accumulate, causing platelet aggregation and microvascular thrombosis. The deficiency may be:
- Acquired (autoantibodies to ADAMTS13)
- Inherited
Hemolytic Uremic Syndrome (HUS)
- Features: Thrombocytopenia + MAHA + renal failure (predominant; less CNS involvement than TTP)
- Pathogenesis: Deficiencies of complement regulatory proteins OR exposure to endothelial-damaging agents (e.g., Shiga toxin from E. coli O157:H7) → platelet activation, aggregation, microvascular thrombosis.
B. Qualitative Platelet Defects
- Acquired: Uremia (uremic toxins impair platelet function), aspirin (irreversibly inhibits COX-1, blocking thromboxane A2), myeloproliferative neoplasms
- Inherited: von Willebrand disease (see below), Glanzmann thrombasthenia (absent GpIIb/IIIa), Bernard-Soulier syndrome (absent GpIb)
III. Coagulation Factor Disorders (Secondary Hemostasis Defects)
Clinical pattern: PT, PTT, or both are prolonged. Petechiae are absent. Hemorrhages occur in areas subject to trauma - classically joints (hemarthroses) and deep soft tissues. Massive hemorrhage after surgery, dental procedures, or severe trauma.
A. von Willebrand Disease (vWD)
- Most common inherited bleeding disorder (~1% of US population)
- Inheritance: Autosomal dominant (most types)
- Mechanism: Mutations in vWF cause:
- Impaired platelet adhesion (vWF bridges platelet GpIb to subendothelial collagen)
- Secondary factor VIII deficiency (vWF normally stabilizes factor VIII in circulation)
- Presentation: Spontaneous bleeding from mucous membranes, excessive bleeding from wounds, menorrhagia. Resembles platelet disorder (mucosal bleeding, petechiae) because primary hemostasis is affected.
- Diagnosis: Ristocetin platelet agglutination test (ristocetin enhances vWF-GpIb binding, causing platelet agglutination - absent/reduced in vWD)
Subtypes:
| Type | Defect | Notes |
|---|---|---|
| Type I (most common) | Quantitative reduction in vWF | Autosomal dominant; mild-moderate bleeding |
| Type IIA | Loss of high-MW multimers (not synthesized) | Functional deficiency |
| Type IIB | Abnormal "hyperfunctional" high-MW multimers rapidly removed | Mild chronic thrombocytopenia from platelet consumption |
| Type III (severe) | Near-complete absence of vWF | Resembles hemophilia (concurrent factor VIII deficiency) |
B. Hemophilia A (Factor VIII Deficiency)
- Most common hereditary cause of serious bleeding
- X-linked recessive - primarily affects males
- ~30% caused by new mutations
- Pathogenesis: Reduced factor VIII activity → impaired intrinsic pathway → PTT prolonged, PT normal
- Severity:
- Severe: factor VIII activity <1% of normal - spontaneous bleeds
- Moderate/Mild: activity 1-30% - bleed with trauma/surgery
- Features:
- Easy bruising, hemorrhage after trauma or surgery
- Hemarthroses (recurrent joint bleeds leading to crippling deformities) - classic
- Deep soft tissue hemorrhages
- Petechiae absent (primary hemostasis intact)
- Diagnosis: Prolonged PTT, corrected by mixing with normal plasma. Specific factor VIII assay confirms.
- Treatment: Recombinant factor VIII infusion. ~15% of severe cases develop factor VIII inhibitor antibodies → treated with bispecific antibody (emicizumab, binds factor IX and X, bypassing factor VIII).
C. Hemophilia B (Factor IX Deficiency - Christmas Disease)
- X-linked recessive, much less common than Hemophilia A
- Clinically indistinguishable from Hemophilia A
- PTT prolonged; diagnosis by specific factor IX assay
- Treated with recombinant factor IX
D. Other Factor Deficiencies
| Factor | Condition | Notes |
|---|---|---|
| Factor VII | Rare autosomal recessive | PT prolonged only |
| Factor XI | Hemophilia C | Mild; common in Ashkenazi Jews |
| Factor XII | Hageman factor deficiency | Prolonged PTT but no bleeding (paradoxically increased thrombosis risk) |
| Vitamin K deficiency | Factors II, VII, IX, X (+ protein C/S) | PT prolonged first; liver disease, newborns, malabsorption, warfarin |
| Liver disease | All factors (except vWF, factor VIII) | PT + PTT both prolonged |
IV. Mixed / Combined Disorders
Disseminated Intravascular Coagulation (DIC)
- "Consumptive coagulopathy" - the most common cause of serious acquired bleeding
- Mechanism: Systemic activation of coagulation → widespread microvascular thrombi → consumption of platelets + coagulation factors → secondary fibrinolysis activation → paradoxical bleeding
Causes:
| Category | Examples |
|---|---|
| Obstetric complications | Abruptio placentae, retained dead fetus, septic abortion, amniotic fluid embolism, eclampsia |
| Infections | Sepsis (gram-negative and gram-positive), meningococcemia, Rocky Mountain spotted fever, malaria |
| Neoplasms | Carcinomas (pancreas, prostate, lung, stomach), acute promyelocytic leukemia (M3) |
| Massive tissue injury | Trauma, burns, extensive surgery, brain trauma |
| Miscellaneous | Snakebite, acute intravascular hemolysis, heat stroke, vasculitis, liver disease, shock |
Pathogenesis:
- Triggered by: (1) release of tissue factor/thromboplastin into circulation, or (2) widespread endothelial damage
- In sepsis: endotoxins stimulate monocytes to express tissue factor and release IL-1/TNF, which upregulate tissue factor on endothelial cells and downregulate thrombomodulin (anti-coagulant)
Morphology:
- Microthrombi in kidneys (most common: fibrin thrombi in glomeruli → renal cortical necrosis), adrenals, brain, heart
- Bilateral adrenal hemorrhagic necrosis (Waterhouse-Friderichsen syndrome in meningococcemia)
- Petechiae and ecchymoses on skin, serosal linings, epicardium, endocardium, lungs, urinary tract
Clinical Features:
- Acute DIC (e.g., obstetric): dominated by bleeding - petechiae, ecchymoses, gut/urinary tract hemorrhage
- Chronic DIC (e.g., adenocarcinoma): dominated by thrombosis - signs of ischemia/organ damage
Lab findings:
- Thrombocytopenia
- PT and PTT both prolonged
- Low fibrinogen
- Elevated fibrin split products (D-dimer) - the hallmark
- Microangiopathic hemolytic anemia (schistocytes on smear)
Treatment: Treat underlying cause; heparin or fresh frozen plasma for acute DIC.
Summary Table
| Disorder | Platelet Count | PT | PTT | Key Features |
|---|---|---|---|---|
| Vascular fragility | Normal | Normal | Normal | Petechiae/ecchymoses, no lab abnormality |
| Thrombocytopenia (ITP) | Low | Normal | Normal | Petechiae, ecchymoses, mucosal bleeding |
| TTP | Low | Normal | Normal | Pentad: MAHA + renal + CNS + fever + thrombocytopenia |
| HUS | Low | Normal | Normal | MAHA + renal failure, Shiga toxin |
| von Willebrand disease | Normal* | Normal | Prolonged* | Mucosal bleeding, menorrhagia, + ristocetin test |
| Hemophilia A & B | Normal | Normal | Prolonged | Hemarthroses, deep bleeds, no petechiae |
| Vitamin K deficiency | Normal | Prolonged | Prolonged | Factors II, VII, IX, X affected |
| DIC | Low | Prolonged | Prolonged | MAHA, D-dimer elevated, petechiae + thrombosis |
*In vWD, PTT is prolonged only if factor VIII is significantly reduced (severe type III); platelet count can be low in type IIB due to consumption.
Source: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; Harrison's Principles of Internal Medicine; The Washington Manual of Medical Therapeutics
Causes of pneumothorax
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Loading Image
Excellent content gathered. Here is the comprehensive answer:
Causes of Pneumothorax
Definition: Pneumothorax is an abnormal collection of air in the pleural space between the parietal and visceral pleura. Normally pleural pressure is subatmospheric; even a small amount of air causes positive pressure that compresses the underlying lung. - Goldman-Cecil Medicine
Classification
Pneumothorax
├── Spontaneous
│ ├── Primary (PSP) - no underlying lung disease
│ └── Secondary (SSP) - underlying lung disease
├── Traumatic
│ ├── Non-iatrogenic (blunt/penetrating)
│ └── Iatrogenic (procedural)
└── Special types
├── Tension pneumothorax
└── Catamenial pneumothorax
1. PRIMARY SPONTANEOUS PNEUMOTHORAX (PSP)
Occurs without any identifiable underlying lung disease or external precipitant.
Mechanism
- Rupture of subpleural apical blebs or bullae (emphysema-like changes found at lung apices)
- Patients with PSP have microscopic structural abnormalities despite an apparently "normal" chest radiograph: distal airways inflammation, "pleural porosity," and emphysema-like changes - Murray & Nadel's Textbook of Respiratory Medicine
- High mechanical stress at lung apices (due to greater negative pleural pressure) promotes bleb formation
Risk Factors
- Tall, thin, young males (ectomorphic body habitus) - 3x more common in men than women; bimodal age distribution with first peak at 15-34 years
- Smoking - significantly increases risk; also increases recurrence risk
- Marfan syndrome - connective tissue laxity predisposes to bleb formation
- Birt-Hogg-Dube syndrome - rare autosomal dominant condition with pulmonary cysts
- Mitral valve prolapse
- Changes in ambient atmospheric pressure
- Genetic predisposition (rare)
2. SECONDARY SPONTANEOUS PNEUMOTHORAX (SSP)
Occurs as a complication of underlying lung disease. Mortality and morbidity are significantly higher than PSP. - Murray & Nadel's
Per the British Thoracic Society (BTS): patients >50 years or with significant smoking history should be classified as SSP even with a normal CXR, given the high likelihood of occult lung pathology.
Causes (Box 63.1 - Rosen's Emergency Medicine):
Airway Disease
- COPD/Emphysema - most common cause in the United States; rupture of emphysematous bullae
- Asthma - air trapping leading to alveolar rupture
- Cystic fibrosis - diffuse bronchiectasis, mucus plugging, and cyst formation
Infections
- Necrotizing bacterial pneumonia / Lung abscess - cavity rupture into pleural space
- Pneumocystis jirovecii pneumonia (PCP) - subpleural cysts and necrosis; notoriously difficult to treat; persistent air leaks; leading cause of pneumothorax in HIV-positive patients
- Tuberculosis - ~7% of HIV patients with pulmonary TB develop pneumothorax; leading cause of SSP in developing countries along with lung abscess
Interstitial Lung Diseases
- Sarcoidosis
- Idiopathic pulmonary fibrosis (IPF)
- Lymphangioleiomyomatosis (LAM) - proliferation of smooth muscle cells causing cystic lung destruction; almost exclusively in women of reproductive age
- Tuberous sclerosis - pulmonary involvement with cysts
- Pneumoconioses
- Eosinophilic granulomatous disease (e.g., Langerhans cell histiocytosis)
Neoplasms
- Primary lung cancers - cavity formation or subpleural tumor rupture
- Pulmonary or pleural metastases
Connective Tissue Diseases
- Marfan syndrome
- Ehlers-Danlos syndrome - defective collagen in lung parenchyma
- Scleroderma
- Rheumatoid arthritis
Miscellaneous
- Pulmonary infarction (from PE)
- Endometriosis - see catamenial pneumothorax below
3. TRAUMATIC PNEUMOTHORAX
A. Non-Iatrogenic Traumatic
Present in approximately 20% of patients with thoracic or polytrauma. - Fishman's Pulmonary Diseases and Disorders
Penetrating trauma:
- Stab wounds, gunshot wounds
- Air enters directly through chest wall OR leaks from a disrupted tracheobronchial tree
- If the chest wall defect is larger than the tracheal diameter (~2 cm), air is preferentially inspired through the chest wall wound ("open/sucking chest wound")
Blunt trauma:
- Rib fracture lacerating the visceral pleura
- Importantly: in ~50% of blunt-trauma pneumothoraces, there are no associated rib fractures - common with blast injuries and high-altitude falls
- Sudden compression can rupture alveoli even without structural rib injury
- Occult pneumothorax - seen on CT but not on initial CXR; majority can be managed conservatively in stable patients
B. Barotrauma (from Mechanical Ventilation)
- Incidence: 4-15% on mechanical ventilation; higher with underlying inflammatory disease (ARDS)
- High ventilatory pressures rupture alveoli - the air tracks along bronchovascular sheaths to the mediastinum (pneumomediastinum) and then into pleural space
- Can rapidly progress to tension pneumothorax
4. IATROGENIC PNEUMOTHORAX
Increasing utilization of invasive procedures is significantly increasing the rate of iatrogenic pneumothorax. - Fishman's Pulmonary Diseases and Disorders
| Procedure | Pneumothorax Rate |
|---|---|
| CT-guided percutaneous fine needle aspiration | 17-26.6% (most common cause) |
| Transbronchial cryobiopsy (TBCB) | 4.8-20% |
| Thoracentesis | ~6% |
| Transbronchial biopsy (TBB) | 1-6% |
| Transbronchial needle aspiration (TBNA) | 1-6% |
| Central line insertion (subclavian) | 0.45-3.1% |
| Navigational bronchoscopy | 4.3% |
| Percutaneous nephrolithotomy | 7% |
| EBUS-TBNA | 0.2% |
| Flexible bronchoscopy | 0.10-0.16% |
| Mechanical ventilation | 4-15% |
Risk factors for iatrogenic pneumothorax: COPD, depth of lesion from pleural surface, lesion size < 2 cm. Real-time ultrasound guidance significantly reduces risk for thoracentesis and central line insertion.
Other iatrogenic causes: tracheostomy, mediastinoscopy, intercostal nerve block, liver biopsy, percutaneous renal biopsy, pacemaker insertion.
5. TENSION PNEUMOTHORAX
Not a separate cause, but a life-threatening complication of any pneumothorax.
The alveolar-pleural defect acts as a one-way valve - air enters the pleural space during inspiration but cannot escape during expiration. Progressive accumulation of intrapleural air causes:
- Mediastinal shift to contralateral side
- Compression of mediastinal venous structures (SVC, IVC)
- Impaired venous return → cardiovascular collapse and death if untreated
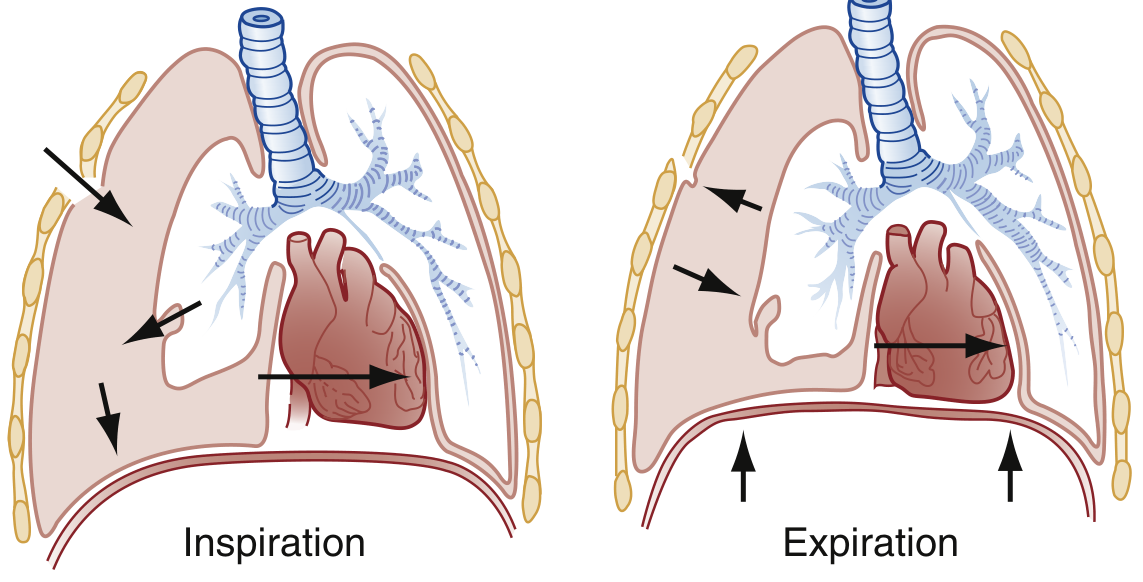
6. CATAMENIAL PNEUMOTHORAX
A special, uncommon type occurring in women of reproductive age (typically 30-35 years).
- Definition: Recurrent pneumothorax associated with pleural or diaphragmatic endometriosis, occurring within 72 hours before or after onset of menstruation
- Almost always right-sided (due to diaphragmatic endometrial implants)
- Mechanism: Rupture of endometrial nodules on the diaphragm or visceral pleura at the time of menstruation → air leaks from peritoneal cavity into pleural space via diaphragmatic defects
- Treatment: Excision/ablation of endometrial lesions, hormonal suppression, repair of diaphragmatic defects
Summary Table
| Type | Key Cause | Population | Notes |
|---|---|---|---|
| Primary Spontaneous | Apical bleb/bulla rupture | Tall, thin, young men | No underlying lung disease |
| Secondary Spontaneous | COPD (USA), TB (developing countries) | Older; existing lung disease | Higher morbidity/mortality than PSP |
| Traumatic - Penetrating | Stab/gunshot wound | Any | Open chest wound - occlude immediately |
| Traumatic - Blunt | Rib fracture, blast injury | Any | 50% have no rib fracture |
| Iatrogenic | Transthoracic needle aspiration, mechanical ventilation | Any (post-procedure) | CT-guided biopsy highest risk |
| Tension | Any of the above (one-way valve mechanism) | Any | Life-threatening emergency |
| Catamenial | Diaphragmatic endometriosis | Women 30-35 years | Right-sided; recurrent; peri-menstrual |
Sources: Rosen's Emergency Medicine; Murray & Nadel's Textbook of Respiratory Medicine; Fishman's Pulmonary Diseases and Disorders; Goldman-Cecil Medicine; The Washington Manual of Medical Therapeutics; Fischer's Mastery of Surgery, 8th Ed.
1 pack year =?
1 pack year = smoking 1 pack (20 cigarettes) per day for 1 year.
Formula
Pack years = (Number of packs smoked per day) × (Number of years of smoking)
Or equivalently:
Pack years = (Number of cigarettes per day ÷ 20) × Years smoked
Examples
| Smoking history | Pack years |
|---|---|
| 1 pack/day × 1 year | 1 pack year |
| 2 packs/day × 10 years | 20 pack years |
| 1/2 pack/day × 20 years | 10 pack years |
| 10 cigarettes/day × 10 years | 5 pack years |
| 40 cigarettes/day × 20 years | 40 pack years |
Clinical Significance
Pack year history is used to:
- Quantify cumulative tobacco exposure in a standardized way
- Lung cancer screening: The USPSTF recommends annual low-dose CT screening for adults aged 50-80 years with a ≥20 pack year history who currently smoke or quit within the past 15 years
- COPD risk stratification - significant COPD risk begins around 10-20 pack years
- Surgical risk assessment - heavy smoking history (>20 pack years) increases perioperative pulmonary complications
- Bladder, head & neck, esophageal cancer risk assessment
A person who smoked 40 cigarettes/day for 30 years has: (40 ÷ 20) × 30 = 60 pack years - considered a very heavy smoker with high lung cancer and COPD risk.
This is a shared conversation. Sign in to Orris to start your own chat.