Theories of colour vision
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Finding Sources
Reading File
I now have rich, authoritative content from Guyton & Hall, Ganong, Kandel's Principles of Neural Science, and Neuroscience: Exploring the Brain. Here is the comprehensive answer:
Theories of Colour Vision
Colour vision has been explained by two historically competing theories - the Trichromatic (Young-Helmholtz) theory and the Opponent Process (Hering) theory - which are now unified into a Zone (stage) theory that describes how each operates at a different level of the visual system.
1. Trichromatic Theory (Young-Helmholtz Theory)
Proposed by: Thomas Young (1802), expanded by Hermann von Helmholtz (1850s).
Core idea: The retina contains three types of cone photoreceptors, each maximally sensitive to one of three primary colours - red, green, and blue. Any colour can be produced by mixing appropriate proportions of these three primary lights.
Photochemical Basis
Each cone contains a different photopigment composed of retinal + photopsin (as opposed to rhodopsin in rods). The three pigments have peak absorbencies at:
| Cone Type | Peak Wavelength | Pigment |
|---|---|---|
| Blue (S-cone) | 445 nm | Blue-sensitive (short-wave) pigment |
| Green (M-cone) | 535 nm | Green-sensitive (middle-wave) pigment |
| Red (L-cone) | 570 nm | Red-sensitive (long-wave) pigment |
The curves overlap significantly, so any single wavelength of light stimulates all three cones to varying degrees. The ratio of stimulation across the three types determines the perceived colour.
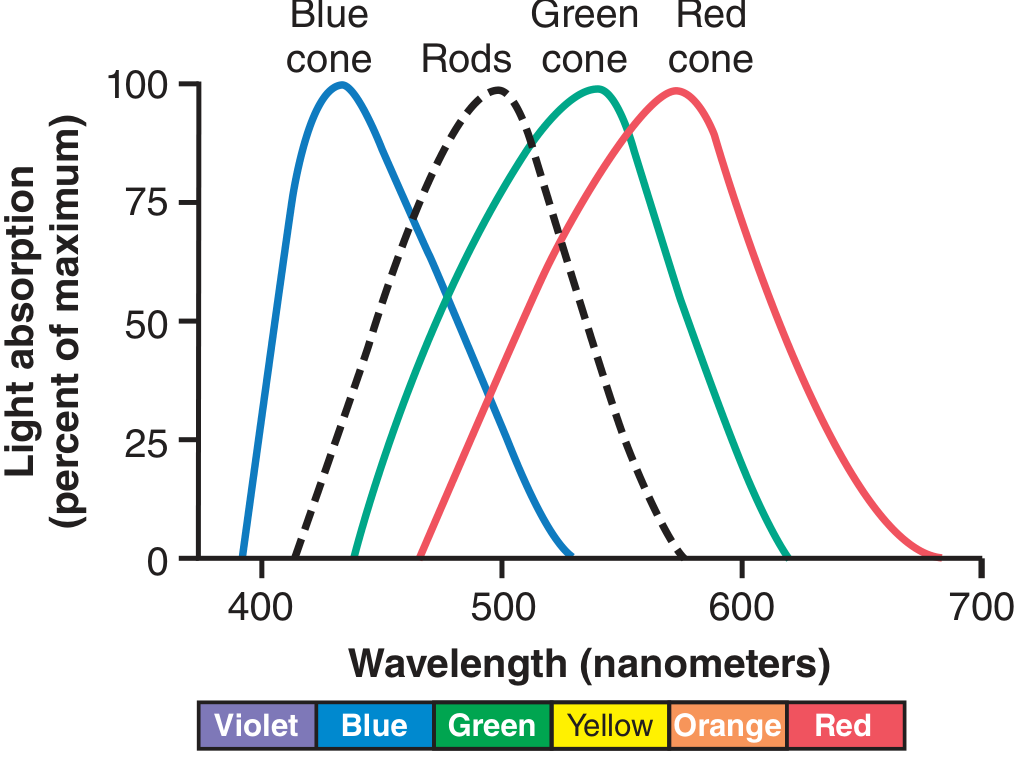
Figure: Light absorption by the pigments of the three colour-receptive cones and rods of the human retina. - Guyton and Hall Textbook of Medical Physiology, p. 637
Key points:
- Red + Green + Blue in equal proportions = sensation of white
- The "red-sensitive" cone actually peaks in the yellow portion of the spectrum but is sensitive enough in the red range to respond at a lower threshold than green cones
- This theory explains why humans are called trichromats
- Early experiments showed any natural colour can be matched by mixing three primary lights - Guyton and Hall Textbook of Medical Physiology, p. 637
Genetic Basis
- Gene for blue (S-cone) pigment: chromosome 7
- Genes for red (L-cone) and green (M-cone) pigments: X chromosome (q arm, in tandem)
- Gene for rhodopsin: chromosome 3
- L and M opsins show 96% amino acid sequence homology with each other, but only ~43% homology with the blue pigment opsin - Ganong's Review of Medical Physiology, 26th Ed., p. 209
2. Opponent Process Theory (Hering's Theory)
Proposed by: Ewald Hering (1878).
Core idea: Colour vision is processed by three opponent channels that respond in mutually inhibitory (push-pull) fashion:
| Channel | Stimulated by | Inhibited by |
|---|---|---|
| Red-Green (r-g) | Red light | Green light |
| Yellow-Blue (y-b) | Yellow light | Blue light |
| White-Black (w-bk) | White (all wavelengths) | Black (no light) |
This elegantly explains phenomena the trichromatic theory cannot:
- Why we never perceive "reddish-green" or "yellowish-blue" - these are mutually exclusive opposites
- Colour afterimages - staring at red then looking at white produces a green afterimage (opponent rebound)
- Simultaneous colour contrast - a colour appears different depending on surrounding colours
Neural Basis (Modern Validation)
Hering's model was later validated by electrophysiology. Opponent processes operate at the ganglion cell and LGN level:
- P-type (parvocellular) ganglion cells - In the central retina, each P-cell receives input from a single L or M cone in its centre and antagonistic surround input:
- L-ON cell: depolarised by red, hyperpolarised by green
- M-ON cell: depolarised by green, hyperpolarised by red
- Small bistratified ganglion cells - receive S-cone (blue) excitation and L+M-cone (yellow) inhibition: depolarised by blue, hyperpolarised by yellow
Three parallel pathways project from retina through the LGN to primary visual cortex (V1):
- Red-Green pathway - signals differences between L- and M-cone responses
- Blue-Yellow pathway - signals differences between S-cone and the sum of L+M cone responses
- Luminance pathway - signals the sum of L+M cone responses (achromatic)
These pathways project to the blobs and deep layer 4C of V1, then onward to area V8 (V4) of the visual cortex, which is uniquely responsible for the conscious sensation of colour. Damage to V8 causes achromatopsia (loss of colour vision) - Ganong's Review of Medical Physiology, 26th Ed.; Eric Kandel - Principles of Neural Science, 6th Ed., p. 585-586
3. Zone (Stage) Theory - The Unified Framework
The Zone theory, developed largely by Müller and later von Kries, reconciles both theories by placing each at a different stage of visual processing:
| Zone | Location | Process | Theory Applicable |
|---|---|---|---|
| Zone I (Receptor level) | Cones in retina | Trichromatic absorption by S, M, L cones | Young-Helmholtz |
| Zone II (Neural coding) | Bipolar cells, ganglion cells, LGN | Opponent-process coding via centre-surround antagonism | Hering |
| Zone III (Cortical) | V1, V8 | Complex colour processing, colour constancy | Both |
This two-stage model is now the accepted framework:
- Stage 1 (retinal): three cone types absorb light trichomatically
- Stage 2 (post-receptoral): retinal circuits convert cone signals into opponent-colour signals
"Color is mediated by ganglion cells that subtract or add input from one type of cone to input from another type." - Ganong's Review of Medical Physiology, 26th Ed., p. 212
Colour Blindness (Defects in Colour Vision)
Understanding these theories also explains colour vision defects:
| Type | Defect | Prevalence |
|---|---|---|
| Trichromats (normal) | All 3 cone pigments present | Normal |
| Anomalous trichromats | One pigment has shifted spectral sensitivity | ~6% of males |
| Dichromats | One cone class absent (protanopia/deuteranopia/tritanopia) | ~2% of males |
| Monochromats | Only one cone class; no colour discrimination | <0.001% |
- Protanopia = absent red (L) cone
- Deuteranopia = absent green (M) cone
- Tritanopia = absent blue (S) cone (rare, autosomal, no sex difference)
Colour blindness is X-linked for red-green defects: 8% of white males, 0.4% of white females are affected - Ganong's Review of Medical Physiology, 26th Ed., p. 209
Summary
| Feature | Trichromatic Theory | Opponent Process Theory |
|---|---|---|
| Proposed by | Young, Helmholtz | Hering |
| Site of action | Cones (retina) | Ganglion cells, LGN |
| Basis | 3 cone types with different peak absorbencies | 3 opponent channels (R-G, Y-B, W-Bk) |
| Explains | Colour mixing, colour blindness types | Afterimages, colour constancy, why no "reddish-green" exists |
| Modern validation | Molecular identification of 3 opsins | Electrophysiology of P-cells and LGN neurons |
Both theories are correct - they describe sequential stages of colour processing. Trichromatic processing at the receptor level feeds into opponent processing at the neural level, and together they account for the full richness of human colour perception.
Hypersensitive reactions
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, authoritative content from Goldman-Cecil Medicine, Janeway's Immunobiology (10e), and Jawetz Medical Microbiology. I have enough for a complete, detailed answer.
Hypersensitivity Reactions
Hypersensitivity reactions are pathological, exaggerated immune responses to antigens (allergens or self-antigens) that damage host tissues. They were classified by Gell and Coombs (1963), later modified by Janeway et al. (2001), into four types (I-IV) based on the immunological mechanism involved.
- Types I, II, and III are antibody-mediated
- Type IV is T cell-mediated
Overview Table
| Feature | Type I | Type II | Type III | Type IV |
|---|---|---|---|---|
| Also called | Immediate / Anaphylactic | Cytotoxic / Cell surface | Immune complex | Delayed-type (DTH) |
| Mediator | IgE | IgG, IgM | IgG (complexes) | T cells (Th1, Th2, Th17, CTL) |
| Antigen | Soluble (allergen) | Cell/matrix-bound | Soluble, circulating | Cell-associated or soluble |
| Effector mechanism | Mast cell degranulation via FcεRI | Complement + FcγR+ phagocytes/NK cells | Complement + FcγR+ cells | Macrophages, eosinophils, cytotoxic T cells |
| Time to reaction | Minutes | Minutes-hours | Hours | 24-72 hours |
| Examples | Anaphylaxis, asthma, hay fever | Hemolytic transfusion reaction, AIHA | Serum sickness, SLE, Arthus reaction | Tuberculin test, contact dermatitis, graft rejection |
(Source: Goldman-Cecil Medicine, Table 36)
Type I - Immediate (IgE-Mediated) Hypersensitivity
Mechanism
Sensitization phase (first exposure):
- Antigen (allergen) is processed by antigen-presenting cells
- Th2 cell response is induced, producing IL-4 and IL-13, which drive B cell class switching to IgE
- IgE binds via its Fc region to FcεRI (high-affinity IgE receptor) on mast cells and basophils - the individual is now sensitized
Effector phase (re-exposure):
4. Allergen crosslinks IgE molecules on the mast cell surface
5. Mast cell degranulation occurs immediately, releasing pre-formed and newly synthesised mediators
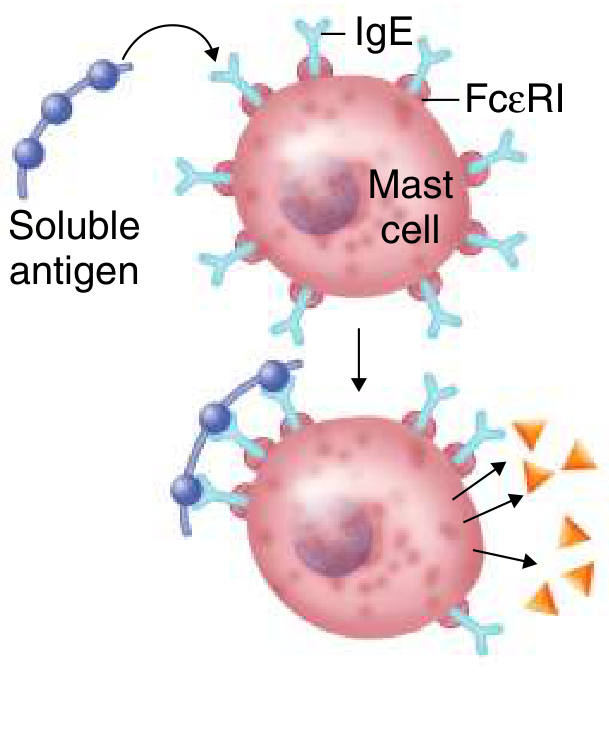
Figure: Type I hypersensitivity - soluble antigen crosslinks IgE on a mast cell bearing FcεRI, triggering degranulation. - Goldman-Cecil Medicine
Mediators Released
Preformed (immediate):
- Histamine - increased vascular permeability, smooth muscle contraction, mucus secretion
- Proteolytic enzymes (tryptase, chymase)
- Heparin
Newly synthesised (minutes-hours):
- Leukotrienes (LTC4, LTD4, LTE4) - bronchoconstriction, mucus production
- Prostaglandins (PGD2) - vasodilation, bronchoconstriction
- Platelet-activating factor (PAF)
- Cytokines: IL-4, IL-5, IL-13, TNF-α, GM-CSF
Biphasic Response
- Immediate phase (within minutes): mast cell mediators - urticaria, bronchoconstriction
- Late phase (2-24 hours): eosinophils, basophils, Th2 cells are recruited; toxic granular proteins (major basic protein, eosinophil peroxidase) cause further tissue damage
Clinical Examples
- Anaphylaxis (systemic) - hypotension, urticaria, angioedema, bronchospasm
- Allergic asthma
- Allergic rhinitis (hay fever)
- Urticaria and angioedema
- Food allergy
Atopy
- Genetic predisposition to IgE-mediated allergy is called atopy
- If both parents are atopic: child has 40-60% chance of developing IgE-mediated allergy
- If neither parent: ~10% risk - Janeway's Immunobiology 10e, p. 682
Type II - Cytotoxic (Antibody-Dependent Cell/Complement Mediated)
Mechanism
- IgG or IgM antibodies are directed against antigens on cell surfaces or extracellular matrix
- Antibody binds antigen on cell surface
- Destruction occurs via:
- Complement activation (C1q → MAC → cell lysis)
- Opsonisation + phagocytosis by macrophages/neutrophils (FcγR-mediated)
- Antibody-Dependent Cellular Cytotoxicity (ADCC) by NK cells bearing FcγRIII (CD16)
In some cases, antibodies to cell-surface receptors can stimulate or block receptor function (receptor-modulating type):
- Graves' disease - anti-TSH receptor antibodies stimulate thyroid (hyperthyroidism)
- Myasthenia gravis - anti-AChR antibodies block neuromuscular junction
Clinical Examples
| Disease | Target antigen | Mechanism |
|---|---|---|
| ABO transfusion reaction | RBC blood group antigens | Complement-mediated lysis |
| Rh haemolytic disease of newborn | Rh antigen on fetal RBCs | IgG ADCC |
| Autoimmune haemolytic anaemia | RBC surface proteins | Complement + ADCC |
| Drug-induced haemolytic anaemia (DIHA) | Drug-coated RBCs | IgG → macrophage clearance in spleen |
| Goodpasture's syndrome | Type IV collagen (glomerular BM) | Complement + neutrophil damage |
| Graves' disease | TSH receptor | Stimulatory antibody |
| Myasthenia gravis | AChR | Blocking antibody |
| Pemphigus vulgaris | Desmoglein (skin desmosomes) | Loss of cell-cell adhesion |
Type III - Immune Complex-Mediated Hypersensitivity
Mechanism
- Circulating soluble antigen-antibody (IgG) complexes form when antigen is in excess
- Small complexes (formed in antigen excess) escape phagocytic clearance and deposit in vessel walls, glomeruli, joints, and skin
- Deposited complexes activate complement (generating C3a, C5a - anaphylatoxins and chemotaxins)
- Neutrophils and macrophages are recruited via FcγR and complement receptors
- These effector cells release proteases and reactive oxygen species → tissue damage
Key Distinguishing Features
- Antigen is soluble (not cell-bound)
- Damage is caused where complexes deposit - often remote from the site of antigen entry
- Complement consumption leads to low serum C3/C4 levels (useful diagnostically)
Local vs. Systemic
Local (Arthus reaction):
- Subcutaneous injection of antigen in an already-immunised (IgG+) individual
- Immune complexes form locally in vessel walls
- Neutrophil infiltration, haemorrhage, necrosis within hours
Systemic (Serum sickness):
- Injection of large amounts of foreign protein (historically horse antiserum)
- Onset: 7-10 days after exposure (time for IgG production)
- Features: fever, rash, urticaria, arthritis, glomerulonephritis
- Resolves when antigen is cleared
Clinical Examples
- Serum sickness (horse antiserum, streptokinase, mouse monoclonal antibodies)
- Systemic lupus erythematosus (SLE) - anti-dsDNA immune complexes deposited in skin, joints, kidneys
- Post-streptococcal glomerulonephritis
- Subacute bacterial endocarditis
- Farmer's lung (inhaled thermophilic actinomycetes antigens)
- Mixed essential cryoglobulinemia (hepatitis C)
Type IV - Delayed-Type Hypersensitivity (DTH) / T Cell-Mediated
Mechanism
- No antibody involved - entirely T cell-dependent
- Time lag of 24-72 hours (hence "delayed") - time needed for sensitised T cells to migrate to site
- Antigen is processed by macrophages/dendritic cells and presented on MHC
Three T cell effector pathways:
IVa - Th1-Mediated (Classic DTH)
- Th1 cells recognise antigen on APCs
- Release IFN-γ → macrophage activation → macrophage-mediated tissue injury
- Also TNF-α, lymphotoxin → endothelial activation, increased permeability
- Example: Tuberculin (Mantoux) test, granuloma formation in tuberculosis, leprosy
IVb - Th2-Mediated
- Th2 cells produce IL-4, IL-5, IL-13, eotaxin → eosinophil recruitment and activation
- Eosinophil degranulation (major basic protein, eosinophil peroxidase) → chronic tissue injury
- Example: Chronic allergic asthma, chronic allergic rhinitis, atopic dermatitis
IVc - Th17-Mediated
- Th17 cells produce IL-17, IL-21, IL-22, GM-CSF → neutrophil recruitment, amplified inflammation
- Implicated in: psoriasis, rheumatoid arthritis, IBD, ankylosing spondylitis
IVd - CTL (CD8+ T Cell)-Mediated
- Cytotoxic CD8+ T cells recognise antigen on MHC class I on target cells
- Direct killing via perforin-granzyme or Fas-FasL
- Example: Contact dermatitis (poison ivy), rejection of virus-infected cells, some allograft rejections
Classic Cellular Hypersensitivity Examples (Janeway's Table)
| Syndrome | Antigen | Consequence |
|---|---|---|
| Delayed-type hypersensitivity (Mantoux) | Tuberculin, mycobacterial proteins | Erythema, induration, cellular infiltrate |
| Contact hypersensitivity | Haptens (DNCB, nickel, chromate, pentadecacatechol/poison ivy) | Epidermal vesicles, intraepidermal abscesses |
| Coeliac disease | α-Gliadin | Villous atrophy, malabsorption |
| Type 1 diabetes mellitus | Islet β-cell antigens | β-cell destruction, insulin deficiency |
| Multiple sclerosis | Myelin basic protein | Demyelination |
Comparison of All Four Types
| Feature | Type I | Type II | Type III | Type IV |
|---|---|---|---|---|
| Immunoglobulin | IgE | IgG / IgM | IgG | None |
| Antigen location | Soluble (free) | Cell/matrix-bound | Soluble (circulating) | Cell-bound or soluble |
| Key cells | Mast cells, eosinophils | NK cells, phagocytes | Neutrophils, phagocytes | Th1/Th2/Th17/CD8+ T cells |
| Complement involved | No (mainly) | Yes | Yes | No |
| Onset | Minutes | Minutes-hours | Hours (local) / days (systemic) | 24-72 hours |
| Passive transfer | With serum (IgE) | With serum (IgG) | With serum (IgG) | With T cells only |
| Prototypic test/disease | Skin prick test, anaphylaxis | Coombs test, transfusion reaction | Arthus reaction, serum sickness | Mantoux (tuberculin) test |
Treatment Principles
| Type | Treatment Approach |
|---|---|
| Type I | Antihistamines, β-agonists, corticosteroids, leukotriene antagonists, omalizumab (anti-IgE), desensitisation/allergen immunotherapy, adrenaline (anaphylaxis) |
| Type II | Remove offending agent, corticosteroids, plasmapheresis, IVIG, specific biologics (e.g. rituximab in AIHA) |
| Type III | Remove antigen source, NSAIDs/corticosteroids, treat underlying disease (e.g. hydroxychloroquine in SLE) |
| Type IV | Topical/systemic corticosteroids, calcineurin inhibitors, biologic agents (e.g. anti-TNF, anti-IL-17 in autoimmune diseases) |
(Sources: Goldman-Cecil Medicine International Edition; Janeway's Immunobiology 10e; Jawetz, Melnick & Adelberg's Medical Microbiology 28e)
This is a shared conversation. Sign in to Orris to start your own chat.