Treatment of patient with sickle cell disease with less episode of crisis complete management
Reading File
Checking Date & Time
Finding Sources
Finding Sources
Searching PubMed
"sickle cell disease"[MeSH Terms] AND "disease management"[MeSH Terms]
Finding Sources
Reading File
Searching PubMed
"sickle cell disease"[MeSH Terms] AND "treatment outcome"[MeSH Terms]
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
I now have all the information needed for a comprehensive answer. Let me compile the complete management guide.
Complete Management of Sickle Cell Disease (Low-Episode / Chronic Phase)
The question focuses on managing a patient with fewer crisis episodes — i.e., a stable patient on long-term disease management. This covers prophylaxis, chronic therapy, disease-modifying agents, monitoring, and complication management.
Pathophysiology (Brief)
Sickle cell disease results from substitution of glutamic acid by valine at the 6th position of the β-globin chain, producing HbS. Under deoxygenation, HbS polymerizes → RBCs sickle → vascular occlusion. Modern understanding implicates multiple mechanisms: activated endothelium, leukocyte adhesion, altered nitric oxide metabolism, hypercoagulability, and ischemia-reperfusion injury — not just polymerization alone.
— Swanson's Family Medicine Review, p. 623
I. DISEASE-MODIFYING / PREVENTIVE PHARMACOTHERAPY
1. Hydroxyurea (First-Line, Cornerstone Therapy)
Mechanism: Oral ribonucleotide reductase inhibitor → increases fetal hemoglobin (HbF) production → HbF dilutes HbS → delays and reduces HbS polymerization → fewer sickled cells → fewer vasoocclusive crises.
Indications: Patients with frequent painful crises, recurrent ACS, or severe anemia. Clinical response takes 3–6 months.
Efficacy: At 12 months, only 75% of treated patients had a painful episode vs. 90% in the placebo group (see graph below).
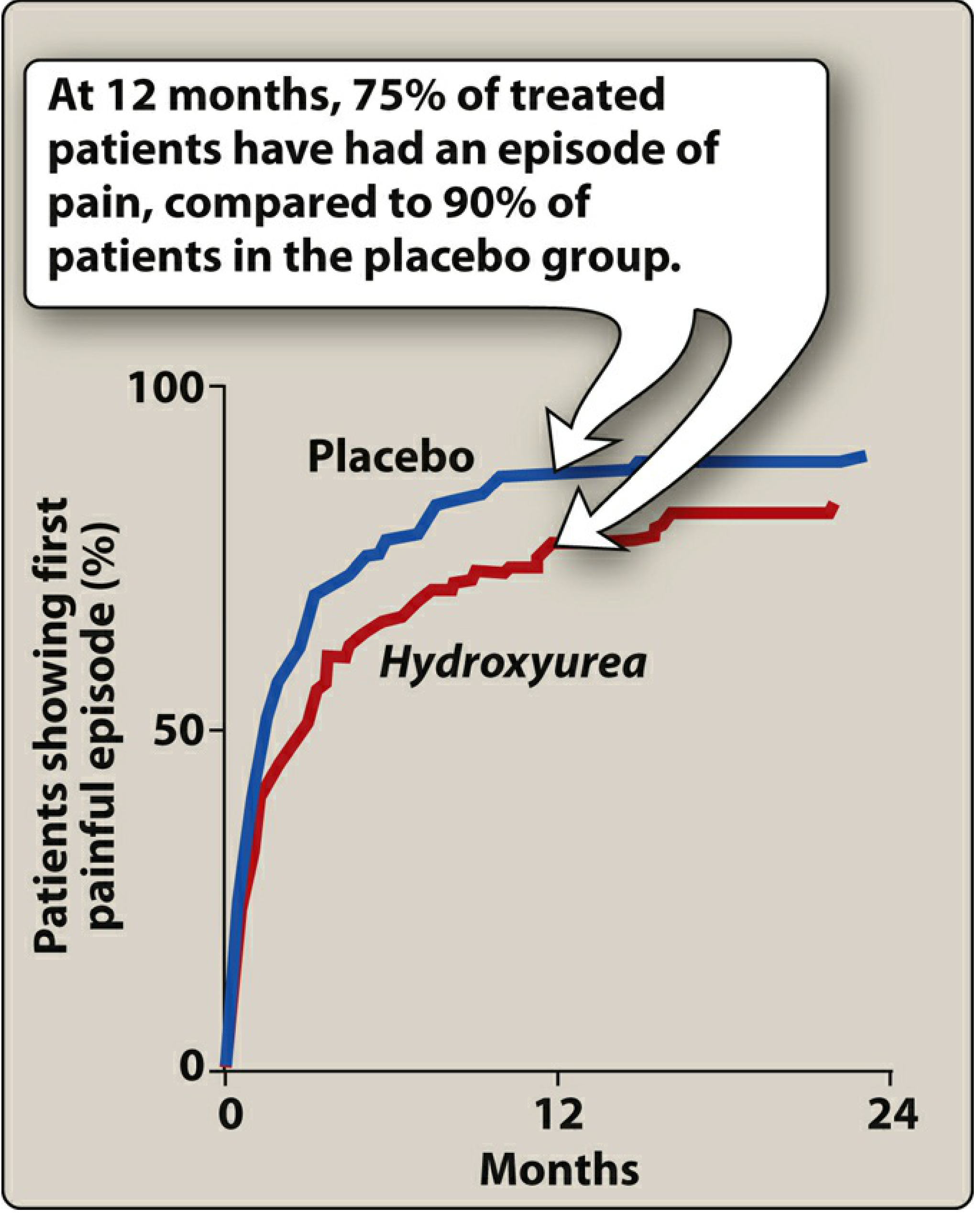
Side effects:
- Bone marrow suppression (monitor CBC)
- Cutaneous vasculitis
- Must be administered under supervision of an experienced provider
Additional uses (off-label): AML, psoriasis, polycythemia vera
— Lippincott Illustrated Reviews: Pharmacology, p. 1483
2. Crizanlizumab (IV Monoclonal Antibody)
Mechanism: Humanized monoclonal antibody → binds P-selectin on activated endothelium and platelets → blocks interactions between endothelial cells, RBCs, platelets, and leukocytes → reduces capillary blockade by sickled cells → fewer crises.
Dosing: IV infusion at weeks 0 and 2, then every 4 weeks
Age: ≥16 years
Adverse effects: Infusion reactions, nausea, arthralgia, back pain, pyrexia
— Lippincott Illustrated Reviews: Pharmacology, p. 1484
3. Voxelotor (Oral, Anti-Sickling)
Mechanism: Binds the alpha chain of HbS → inhibits HbS polymerization by decreasing the concentration of deoxygenated HbS (the form that polymerizes and causes sickling).
Dosing: Once-daily oral dose
Age: ≥12 years
Adverse effects: Headache, diarrhea, GI upset
Drug interactions: Metabolized by CYP3A4 — dose adjustments needed with CYP3A4 inducers/inhibitors
— Lippincott Illustrated Reviews: Pharmacology, p. 1484
4. Folic Acid Supplementation
Mandatory for all patients due to chronic hemolysis causing accelerated folate consumption and risk of aplastic crisis (exacerbated by folate deficiency or parvovirus B19 infection). Daily supplementation is standard.
II. INFECTION PROPHYLAXIS (Critical in Functional Asplenia)
Sickle cell disease causes functional asplenia from repeated splenic infarction → high risk for encapsulated organisms (especially Streptococcus pneumoniae, H. influenzae, N. meningitidis).
| Intervention | Details |
|---|---|
| Penicillin V prophylaxis | Start at 2 months of age; continue until age 5 (unless history of invasive pneumococcal infection or splenectomy — continue longer) |
| Pneumococcal vaccine (PCV13) | Children <2 years; reduces invasive pneumococcal infection |
| Pneumococcal vaccine (PPSV23) | Age ≥2 years in addition to PCV13 |
| Annual influenza vaccine | Due to functional asplenia and immune compromise |
| Meningococcal vaccine | Recommended |
| All standard childhood vaccines | Must be kept up to date |
— Swanson's Family Medicine Review, p. 624
III. STROKE PREVENTION / NEUROLOGIC MONITORING
- Transcranial Doppler (TCD) ultrasonography is recommended for asymptomatic children to screen for elevated blood flow velocity
- If TCD velocity ≥ 200 cm/s → high stroke risk → candidate for chronic transfusion therapy
- Up to 30% of HbSS patients suffer a stroke; recurrence is common
- Extended submandibular approach to TCD can increase sensitivity
IV. TRANSFUSION THERAPY
| Indication | Setting |
|---|---|
| Acute stroke | Exchange transfusion |
| Acute chest syndrome with hypoxia or deterioration | Simple or exchange transfusion |
| Splenic sequestration | Transfusion |
| Acute multiorgan failure | Rapid exchange transfusion |
| Chronic stroke prevention (high TCD velocity) | Chronic transfusion program |
Exchange transfusion improves microvascular perfusion and reduces inflammatory mediators. Monitor HbS concentration before and after exchange.
— Rosen's Emergency Medicine, p. 2964; Miller's Anesthesia, p. 3469
V. MANAGEMENT OF ACUTE VASOOCCLUSIVE CRISIS
When a patient presents with pain crisis:
- Analgesia — Aggressive pain management with opioid analgesics (morphine/hydromorphone) titrated to effect; do NOT undertreat
- IV hydration — Correct dehydration; avoid fluid overload
- Oxygen — If SpO₂ <92% or PaO₂ <70 mmHg
- NSAIDS — As adjunct analgesics
- Incentive spirometry — Prevent hypoventilation and secondary ACS
- Avoid triggers — Cold, dehydration, infection, stress
VI. MANAGEMENT OF ACUTE CHEST SYNDROME (ACS)
ACS = most common cause of death in sickle cell disease; 30% of HbSS patients have ≥1 episode.
Diagnosis: New infiltrate on CXR + fever, chest pain, dyspnea, cough, hypoxemia
Triggers: Infection, fat embolism, rib/sternal/thoracic vertebral infarcts → hypoventilation
Treatment:
- Hematology/pulmonology consultation
- Supplemental O₂ (target SpO₂ ≥92%)
- Incentive spirometry while awake
- Broad-spectrum antibiotics (include atypical organism coverage — Mycoplasma, Chlamydia)
- Careful fluid management (intake/output monitoring)
- Bronchodilators if bronchospasm present
- Aggressive pain management with respiratory monitoring
- Transfusion therapy for hypoxemia or deterioration (exchange if severe)
— Swanson's Family Medicine Review, p. 624
VII. CHRONIC COMPLICATIONS AND MONITORING
| Organ System | Complication | Management |
|---|---|---|
| Bone | Avascular necrosis (femoral head) | Orthopedic referral; analgesics; possible joint replacement |
| Eyes | Proliferative retinopathy, vitreous hemorrhage, retinal detachment | Annual ophthalmology screening |
| Kidneys | Papillary necrosis, hematuria, glomerulonephropathy | Nephrology; ACE inhibitors if proteinuria |
| Liver/Gallbladder | Cholelithiasis (bilirubin stones from chronic hemolysis) | Surgical referral if symptomatic |
| Heart | High-output cardiac failure (from chronic anemia/hypoxemia) | Manage anemia; treat CHF |
| Lungs | Pulmonary hypertension (up to 30% of adults) | Echocardiography screening; cardiology referral |
| CNS | Stroke, TIA | TCD screening; transfusion program |
| Genitourinary | Priapism, hematuria, renal failure | Hydration, oxygenation, pain management; urology |
VIII. CURATIVE THERAPY
| Option | Details |
|---|---|
| Allogeneic hematopoietic stem cell transplant (HSCT) | Only established cure; best outcomes in young patients with HLA-matched sibling donor; risk of graft-vs-host disease |
| Gene therapy | Emerging — lentiviral vector-based HbF induction or gene editing (CRISPR); shows promise in clinical trials |
IX. PREOPERATIVE CONSIDERATIONS
- Do NOT operate during a crisis unless emergent
- Continue routine medications (including hydroxyurea)
- Hydroxyurea stimulates HbF production and reduces operative risk
- Consider preoperative transfusion to reduce HbS percentage
— Miller's Anesthesia, 10e, p. 1339
Summary Table: Chronic Preventive Regimen for a Patient with Few Episodes
| Intervention | Purpose |
|---|---|
| Hydroxyurea | ↑ HbF, ↓ crisis frequency |
| Crizanlizumab (≥16 yr) | Anti-P-selectin, ↓ adhesion and crises |
| Voxelotor (≥12 yr) | Anti-polymerization |
| Folic acid daily | Prevent aplastic crisis |
| Penicillin V (children <5 yr) | Prevent pneumococcal sepsis |
| Vaccines (pneumococcal, influenza, meningococcal) | Infection prevention |
| TCD screening (children) | Stroke risk stratification |
| Annual ophthalmology | Retinopathy surveillance |
| Echocardiography | Pulmonary HTN screening |
| Renal function monitoring | Early nephropathy detection |
Sources:
- Lippincott Illustrated Reviews: Pharmacology — Hydroxyurea, Crizanlizumab, Voxelotor
- Swanson's Family Medicine Review — Clinical management, vaccination, ACS
- Rosen's Emergency Medicine — Acute crisis management, transfusion indications
- Miller's Anesthesia, 10e — Perioperative management
Dosage of hydroxylurea
Checking Date & Time
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
The Harriet Lane page matched carbamazepine, not hydroxyurea. I now have all needed dosing info from two authoritative pulmonary textbooks. Here is the complete answer:
Hydroxyurea Dosing in Sickle Cell Disease
Standard Adult & Pediatric Dosing
| Parameter | Dose |
|---|---|
| Starting dose | 15 mg/kg/day orally, once daily |
| Titration | Increase gradually every 8–12 weeks based on response and tolerability |
| Maximum dose | 35 mg/kg/day |
Hydroxyurea is started at a dose of 15 mg/kg/day and titrated up to a maximum of 35 mg/kg/day. — Fishman's Pulmonary Diseases & Disorders; Murray & Nadel's Textbook of Respiratory Medicine
Renal Adjustment
- The above dosing applies to patients with creatinine < 1 mg/dL
- Patients with renal dysfunction do not tolerate hydroxyurea's myelosuppressant effects well
- In renal impairment: reduce dose or add erythropoietin to the regimen as an adjunct
Titration Principles
- Start at 15 mg/kg/day (once daily, oral)
- Check CBC every 4 weeks during titration
- Increase by 5 mg/kg/day increments every 8–12 weeks if:
- No significant myelosuppression
- Adequate HbF response not yet achieved
- Target: maximum tolerated dose up to 35 mg/kg/day
- Clinical response (↑ HbF, ↓ crisis frequency) takes 3–6 months
Monitoring Parameters (CBC Thresholds — Hold/Reduce Dose)
| Lab Parameter | Hold/Reduce If |
|---|---|
| Neutrophils (ANC) | < 2,000/µL |
| Platelets | < 80,000/µL |
| Hemoglobin | < 4.5 g/dL (or rapid drop) |
| Reticulocytes | < 80,000/µL (if Hgb < 9 g/dL) |
If any threshold is crossed → withhold hydroxyurea → recheck CBC in 2 weeks → restart at a lower dose once counts recover.
Key Drug Interactions
| Drug | Interaction |
|---|---|
| HIV antiretrovirals (e.g., didanosine, stavudine) | Hydroxyurea ↓ CD4 counts; increased toxicity |
| Salicylates/NSAIDs | Increased bleeding risk |
| Probenecid | ↑ uric acid |
Special Populations
- Children: Now recommended from infancy through adulthood (studies in very young children ongoing); same weight-based dosing (15–35 mg/kg/day)
- Pregnancy: Teratogenic — contraindicated; use effective contraception
- Renal impairment: Dose reduce; consider adding erythropoietin
Key Adverse Effects to Monitor
| Effect | Detail |
|---|---|
| Myelosuppression | Most important; monitor CBC regularly |
| Skin ulcers | Especially leg ulcers |
| Secondary leukemia | Long-term risk (low but real) |
| Elevated liver enzymes | Monitor LFTs |
| GI effects | Nausea, vomiting |
Bottom line: Start at 15 mg/kg/day PO once daily, titrate up by ~5 mg/kg/day every 8–12 weeks to a maximum of 35 mg/kg/day, guided by CBC monitoring and clinical response. Allow 3–6 months to assess full therapeutic effect.
— Fishman's Pulmonary Diseases and Disorders; Murray & Nadel's Textbook of Respiratory Medicine, p. 4656; Lippincott Illustrated Reviews: Pharmacology; Katzung's Basic and Clinical Pharmacology, 16e

Should I start it with this patient ???
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
I have enough context from the prior conversation and the HPLC report itself to give a clinically precise answer. Let me analyze the report carefully.
Interpreting This HPLC Report
This is a Bio-Rad D-10 HPLC (HbA2/F method) — a high-performance liquid chromatography used for hemoglobin variant analysis.
Result Summary
| Peak | Area % | Interpretation |
|---|---|---|
| HbF | 23.6% | Very elevated (normal adult <1%) |
| HbA0 | 6.2% | Small amount of normal adult Hb |
| HbA2 | 4.6% | Slightly elevated (borderline) |
| S-Window | 63.0% | HbS — dominant hemoglobin |
| HbA1a/A1b | 4.6% | Glycated fractions |
Diagnosis
The pattern — HbS 63% + HbF 23.6% + HbA 6.2% — is consistent with Sickle Cell Disease (HbSS) in a patient who already has a significantly elevated baseline HbF level (23.6%).
This is an unusually high HbF for an untreated HbSS patient and may represent:
- A patient with a hereditary persistence of fetal hemoglobin (HPFH) variant alongside HbS → milder phenotype
- Or a patient already receiving some form of HbF-inducing therapy
Should You Start Hydroxyurea?
The Key Issue: HbF is Already 23.6%
Hydroxyurea works by raising HbF. The therapeutic target with hydroxyurea is typically an HbF rise to 15–25%, which is associated with reduced crisis frequency and improved survival.
This patient's HbF is already at 23.6% — at the upper end of the therapeutic target range.
Clinical Decision Framework
| Consideration | This Patient |
|---|---|
| HbF level | 23.6% — already in therapeutic range |
| HbS level | 63% — still significantly high |
| HbA (normal) | Only 6.2% — confirms symptomatic disease |
| Crisis frequency | Stated as "less episodes" — mild/stable phenotype |
Answer: It Depends on Clinical Severity — but Caution is Warranted
Arguments AGAINST starting hydroxyurea now:
- HbF is already 23.6% — the patient may be experiencing natural/genetic HbF elevation (HPFH co-inheritance), explaining the fewer crises
- Adding hydroxyurea when HbF is already high offers diminishing additional benefit and adds myelosuppression risk
- The patient already has a mild phenotype (fewer episodes) — consistent with the protective effect of high HbF
- Starting hydroxyurea without a clear indication (few crises, already high HbF) is not standard practice
Arguments FOR starting hydroxyurea:
- HbS is still 63% — there is residual sickling risk
- Hydroxyurea has additional benefits beyond HbF induction: reduces leukocyte adhesion, nitric oxide effects, reduces neutrophilia
- If this HbF is naturally high (HPFH), hydroxyurea may push it even higher (>25–30%), providing further protection
Clinical Recommendation
Do NOT rush to start hydroxyurea in this patient based on this HPLC alone.
Before deciding:
- Confirm the diagnosis — rule out HbS/HPFH vs. true HbSS with naturally elevated HbF (repeat testing, family studies, molecular genetics)
- Assess crisis history carefully — if truly ≤2 moderate crises/year, the high HbF may be providing sufficient protection
- If crisis frequency is significant (≥3/year, hospitalizations, ACS episodes, severe anemia) → start hydroxyurea at 15 mg/kg/day despite the high HbF, as it may push HbF higher still and offer additional non-HbF benefits
- Check CBC, renal function, and LFTs before initiating
Bottom line: This HPLC shows a patient whose high HbF (23.6%) is likely already protecting them — explaining the fewer crises. Hydroxyurea is not urgently indicated based on this result alone, but the final decision rests on clinical severity, crisis frequency, and end-organ damage — not the HPLC in isolation.
How to rule out persistent HBF condition form sickle cell disease??
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
I now have comprehensive information from four authoritative textbooks. Let me compile the complete answer.
How to Distinguish HPFH from Sickle Cell Disease with Elevated HbF
Why This Matters for Your Patient
Your patient's HPLC showed: **HbS 63% | HbF 23.6% | HbA 6.2% | HbA2 4.6%
This pattern fits two possible diagnoses:
- HbSS + co-inherited HPFH (HbSS-HPFH) — milder, pancellular HbF distribution
- HbSS with naturally/stress-elevated HbF — still significant sickling disease
The distinction changes management completely.
Step-by-Step Approach to Rule Out HPFH
Step 1 — Peripheral Blood Indices (CBC + Film)
| Feature | HPFH | HbSS (elevated HbF only) |
|---|---|---|
| MCV / MCH | Normal (normochromic, normocytic) | Normal or mildly reduced |
| Hemoglobin level | Normal or near-normal | Low (7–10 g/dL) |
| RBC morphology | No sickled cells (or very few) | Sickled cells, target cells |
| Anemia | Absent | Present |
"A patient with significantly elevated HbF and reduced HbA2 is suspected to have HPFH if the red cell indices are normal" — Henry's Clinical Diagnosis and Management by Laboratory Methods
"Hb, MCV, and MCH are within the reference intervals. This condition is clinically innocuous and asymptomatic." — Tietz Textbook of Laboratory Medicine, 7th Edition
Key point: In pure HPFH, the patient is not anemic and indices are normal. Your patient's clinical picture (sickle cell disease with crises) already suggests this is SCD + elevated HbF, not pure HPFH.
Step 2 — HbA2 Level (Critical Discriminator)
| Finding | HPFH | δβ-thalassemia / SCD alone |
|---|---|---|
| HbA2 | Low/normal (1.0–2.1%) | Normal to elevated (>3.5%) |
In deletional pancellular HPFH: HbA2 is characteristically decreased (1–2.1%) because the δ-gene complex is deleted along with the β-gene.
Your patient's HbA2 = 4.6% — this is elevated, which argues AGAINST pure HPFH and is more consistent with HbSS + β-thalassemia trait or simply HbSS with reactive HbF elevation.
"In the heterozygote (deletional HPFH), HbF is 15–30% and HbA2 is decreased at 1–2.1%" — Henry's Clinical Diagnosis and Management by Laboratory Methods
Step 3 — Kleihauer-Betke Test (Acid Elution) / Flow Cytometry for F-cells
This is the most important single test to differentiate HPFH from other causes of elevated HbF.
| Distribution Pattern | Diagnosis |
|---|---|
| Pancellular — HbF evenly distributed in ALL red cells | HPFH |
| Heterocellular — HbF in some cells, absent in others | SCD, β-thalassemia, stress erythropoiesis |
Method:
- Kleihauer-Betke acid elution stain — HbF-containing cells resist acid elution (stain pink); HbA cells become ghost cells
- Flow cytometry with anti-γ-globin monoclonal antibody — more accurate, especially at low HbF concentrations; quantifies F-cell percentage
"HPFH is characterized by HbF in a pancellular distribution... Combined sickle cell & β-thalassemia also shows HbS, HbF, and HbA2 bands, but with a heterocellular distribution. This is clinically more severe than SS-HPFH." — Quick Compendium of Clinical Pathology, 5th Edition
"The nondeletional HPFH increase in HbF is distributed heterogeneously (heterocellular) among red cells. The HbF distribution is also heterocellular in β-thalassemia, while it is pancellular in deletional HPFH." — Tietz Textbook of Laboratory Medicine, 7th Edition
Step 4 — Family Studies / Parental Testing
- Test both parents with HPLC hemoglobin analysis
- HPFH is autosomal — if one parent has elevated HbF with normal indices → supports HPFH co-inheritance
- Also helps identify coinherited α-thalassemia (common in African Americans, modifies phenotype)
Step 5 — Molecular / DNA Analysis (Definitive)
| Test | What It Detects |
|---|---|
| β-globin gene cluster deletion analysis | Identifies specific HPFH deletions (Black, Ghanaian, Indian, Italian forms) |
| γ-globin promoter sequencing | Nondeletional HPFH point mutations |
| BCL11A / KLF1 variant testing | Quantitative trait loci for HbF regulation (heterocellular forms) |
| Gap-PCR / MLPA | Detects large deletions in the β-globin locus |
"Several deletional variants of HPFH have been described, including Greek, Indian, Italian, Thai, Corfu, and black forms." — Tietz Textbook of Laboratory Medicine, 7th Edition
Summary Diagnostic Algorithm
Elevated HbF in a sickle cell patient
↓
1. CBC + film → Normal indices + no sickled cells? → Favors HPFH
↓
2. HbA2 level → Low (<2%)? → Favors deletional HPFH
High (>3.5%)? → Favors SCD ± β-thal or reactive elevation
↓
3. Kleihauer-Betke / Flow cytometry
→ Pancellular HbF → HPFH confirmed
→ Heterocellular HbF → SCD with reactive HbF / β-thalassemia
↓
4. Family studies (test parents)
↓
5. Molecular genetics (if needed for definitive confirmation)
Applying This to Your Patient
| Feature | Your Patient | Expected in HPFH |
|---|---|---|
| HbF | 23.6% | 15–30% ✓ |
| HbA2 | 4.6% | Should be LOW (1–2.1%) ✗ |
| HbA (normal) | 6.2% present | Should be absent in homozygous HPFH ✗ |
| Clinical crises | Present | None in pure HPFH ✗ |
The elevated HbA2 (4.6%) and presence of crises strongly argue against pure HPFH. The most likely interpretation remains HbSS with a naturally high HbF level (possibly due to heterocellular HbF quantitative trait loci — a common finding in African Americans).
The confirmatory test to order next: Kleihauer-Betke stain or flow cytometric F-cell quantification — if heterocellular → HbSS with reactive HbF; if pancellular → HPFH co-inheritance.
Sources: Henry's Clinical Diagnosis and Management by Laboratory Methods; Quick Compendium of Clinical Pathology, 5th ed.; Tietz Textbook of Laboratory Medicine, 7th ed.; Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.