Transverse myelitis
Reading File
Finding Sources
Searching PubMed
"transverse myelitis"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Let me compile the full answer.
Transverse Myelitis
Definition
Transverse myelitis (TM) is an acute or subacute inflammatory-demyelinating lesion of the spinal cord, presumed to be autoimmune in origin, that interrupts ascending and descending pathways at a given cord level. The term "transverse" implies involvement of the full cross-sectional area - though in practice this is often incomplete, particularly in MS-associated cases. It is rare, affecting approximately 1 in 100,000 people, and is usually monophasic. - Goldman-Cecil Medicine, p. 3995
Etiology and Associations
TM is heterogeneous - it can be:
| Category | Examples |
|---|---|
| Idiopathic | No cause found in ~30% of cases |
| Post-infectious | Follows viral illness in ~30%; termed postinfectious myelitis |
| Demyelinating disease | Multiple sclerosis (MS), Neuromyelitis Optica Spectrum Disorder (NMOSD) |
| Autoimmune / CTD | SLE, Sjögren syndrome, antiphospholipid antibody syndrome, vasculitis |
| Infectious | Direct viral/bacterial cord invasion |
| Vascular | Dural AV fistula, spinal cord infarction |
| Post-vaccination | Rare, described after various vaccines |
Key antibody associations: anti-AQP4 (aquaporin-4) antibodies are strongly associated with NMOSD-related TM, and anti-MOG (myelin oligodendrocyte glycoprotein) antibodies are seen in MOG antibody disorder. - Rosen's Emergency Medicine, p. 1508 | Goldman-Cecil, p. 3995
Pathophysiology
- The cord lesion involves inflammatory cell infiltration and demyelination, disrupting both ascending sensory pathways (spinothalamic tract, posterior columns) and descending motor pathways (corticospinal tract), as well as autonomic fibers.
- When the lesion spans 3 or more vertebral body segments rostrocaudally, it is called Longitudinally Extensive Transverse Myelitis (LETM) - a hallmark of NMOSD.
- The thoracic cord is affected in 60-70% of cases; cervical cord involvement is less common.
- Progression is rapid: 66% reach maximal deficit within 24 hours, though progression can occur over days to weeks. - Rosen's Emergency Medicine, p. 1508
Clinical Features
Classic triad:
- Motor - bilateral weakness progressing to paraparesis/paraplegia; hypertonia, hyperreflexia, clonus, and Babinski signs once spinal shock resolves
- Sensory - distinct sensory level on the trunk; ascending paresthesias; loss of deep sensation (vibration, proprioception) in the feet
- Autonomic - bladder/bowel dysfunction (urinary and fecal retention or incontinence); sexual dysfunction; in cervical/high thoracic lesions: hyper- or hypotension, tachy- or bradycardia
Additional features:
-
Back or neck pain is common at onset
-
Low-grade fever may be present
-
In MS-associated TM, deficits are often asymmetrical and incomplete (partial TM) - full paraplegia and complete sensory loss are unusual
-
In NMOSD-associated TM, the picture is typically fulminant and complete
-
Adams and Victor's Neurology, p. 925 | Rosen's Emergency Medicine, p. 1508 | Tintinalli's Emergency Medicine, p. 1929
Diagnosis
MRI (Investigation of Choice)
MRI with gadolinium enhancement is the gold standard. Findings include:
- T2 hyperintensity in the spinal cord at the level of involvement
- Cord swelling/expansion in acute phase
- Gadolinium enhancement (patchy or ring)
- LETM (>3 vertebral segments) favors NMOSD over MS
In MS, the cord lesion tends to be short (< 2 vertebral segments), asymmetric, and peripheral.
The MRI image below shows MS findings including periventricular white matter plaques and cervical cord lesions for comparison:
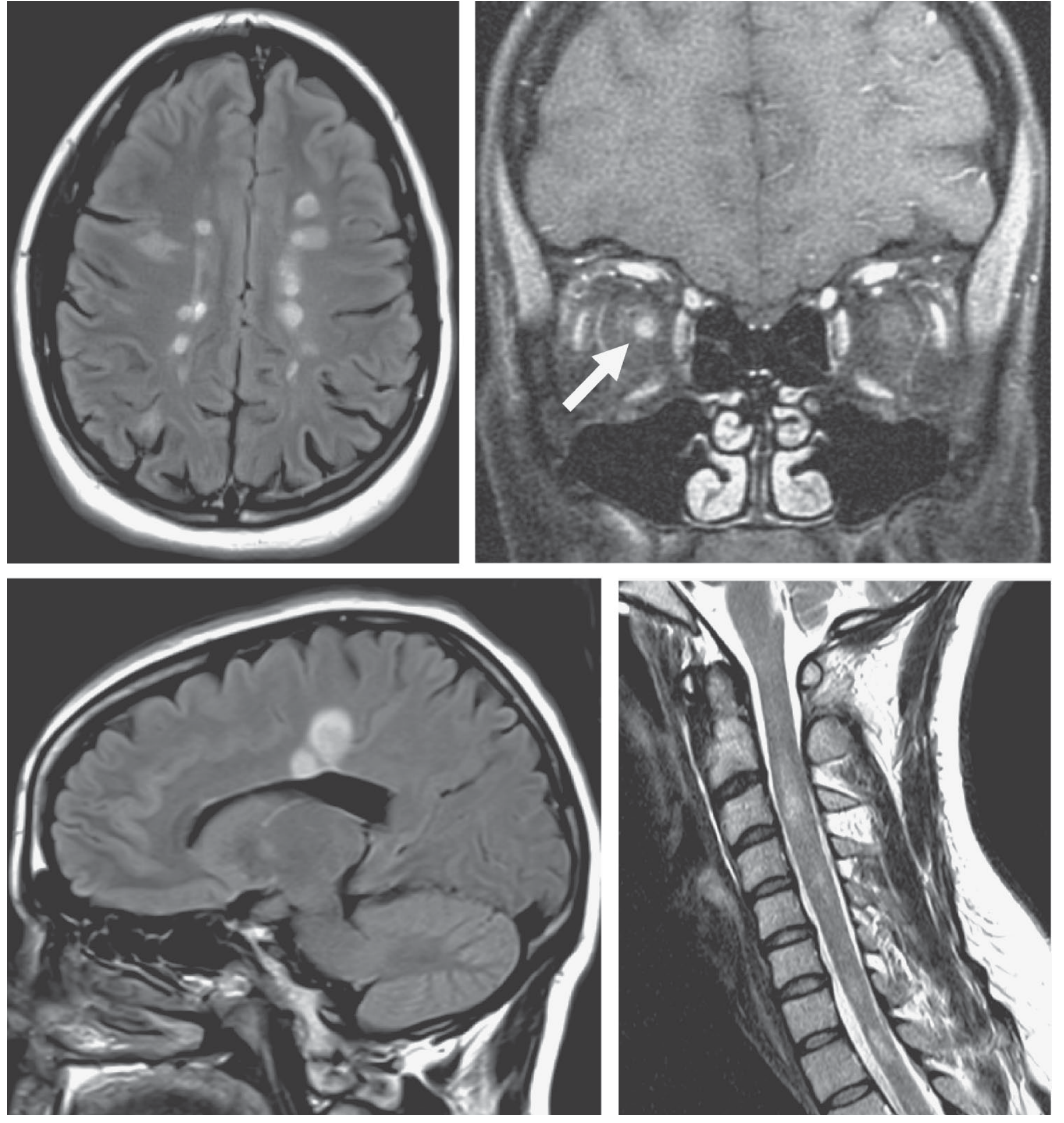
CSF
- Normal in 40% of cases
- Remaining 60%: mild lymphocytosis and elevated protein
- Oligoclonal bands favor MS
- Note: CSF findings may lag the clinical presentation
Serology
-
Anti-AQP4-IgG (NMO-IgG): specific for NMOSD
-
Anti-MOG-IgG: MOG antibody disorder
-
ANA, anti-dsDNA, antiphospholipid antibodies: for CTD work-up
-
Infectious serologies as clinically indicated
-
Rosen's Emergency Medicine, p. 1508 | Bradley and Daroff's Neurology, p. 472
Differential Diagnosis
| Condition | Key distinguishing features |
|---|---|
| Spinal epidural abscess | Fever + severe back pain + focal tenderness; prior procedure/IVDU |
| Spinal cord compression | Disk, tumor, hematoma; gradual onset; MRI shows extrinsic compression |
| MS | Short, asymmetric, incomplete cord lesion; brain MRI shows additional plaques |
| NMOSD | LETM, bilateral optic neuritis, AQP4-Ab positive |
| Spinal cord infarction | Sudden onset, vascular risk factors; anterior cord syndrome pattern |
| Spinal epidural hematoma | Anticoagulation history; acute-onset back pain |
| Vitamin B12 deficiency | Subacute combined degeneration; posterior column + corticospinal tracts |
| Dural AV fistula | Progressive myelopathy; T2 changes + flow voids on MRI |
- Rosen's Emergency Medicine, p. 1508 | Bradley and Daroff's Neurology, p. 472
Treatment
First-line: IV Corticosteroids
- Methylprednisolone 1000 mg IV daily for 3-5 days is the standard approach
- Note: Evidence base is limited; most benefit is inferred from MS trials
Steroid-refractory or severe cases:
- Plasma exchange (PLEX) - particularly effective in NMOSD and aggressive post-infectious TM
- IVIG - an alternative, especially in post-infectious cases
- Cyclophosphamide - for refractory cases
NMOSD-specific preventive therapy (to reduce relapses):
- Eculizumab (terminal complement inhibitor, 900 mg IV weekly x4, then 1200 mg q2 weeks)
- Satralizumab (anti-IL-6R, 120 mg SC at 0, 2, 4 weeks, then q4 weeks)
- Tocilizumab (anti-IL-6R, 8 mg/kg IV q4 weeks)
- Older options: Rituximab (1000 mg IV x2, 2 weeks apart, q6 months) or Azathioprine (3 mg/kg/day)
All patients require neurologic consultation and hospitalization.
- Goldman-Cecil Medicine, p. 3995
Prognosis
-
Outcome varies widely from complete recovery to permanent paralysis
-
Recovery typically begins within 1-3 months; maximal improvement by 3-6 months
-
Outcomes (Goldman-Cecil / Rosen's):
- ~30% "good" recovery
- ~25% "fair" recovery
- ~30% "poor" outcome (remain significantly disabled)
- ~15% mortality at 5 years
-
Prognosis is worse than in MS - significant recovery in fewer than 50%
-
Risk of developing MS: fewer than half develop evidence of additional demyelinating disease within 5 years; cumulative risk is similar after TM vs. optic neuritis (Beck et al., 2002)
-
Adams and Victor's Neurology, p. 926 | Rosen's Emergency Medicine, p. 1508 | Goldman-Cecil Medicine, p. 3995
Key Distinction: Partial vs. Complete TM
| Feature | Partial (incomplete) TM | Complete TM |
|---|---|---|
| Motor | Asymmetric weakness | Bilateral paraplegia |
| Sensory | Incomplete level | Complete sensory level |
| Association | More often MS | More often NMOSD or idiopathic |
| Lesion length | Short (<2 segments) | LETM (≥3 segments) |
| Prognosis | Better | Worse |
Summary
Transverse myelitis presents with the triad of motor weakness, sensory level, and autonomic dysfunction. The primary workup is MRI with gadolinium to exclude compressive lesions and characterize cord signal change. Serology for AQP4 and MOG antibodies is essential to identify NMOSD/MOGAD. Treatment is IV methylprednisolone, with PLEX or IVIG for refractory cases. Long-term immunosuppression is required in NMOSD to prevent relapses.
Bells palsy
Finding Sources
Searching PubMed
"Bell palsy"[MeSH Terms]
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have excellent, comprehensive information from Harrison's, Adams & Victor's, Goldman-Cecil, and Scott-Brown's. Let me compile the full answer.
Bell's Palsy
Definition
Bell's palsy is an acute, idiopathic, unilateral lower motor neuron (LMN) facial paralysis - a diagnosis of exclusion. It is the most common cause of facial paralysis, affecting ~23-25 per 100,000 people annually, which translates to approximately 1 in 60 persons over a lifetime. - Harrison's, p. 3597 | Adams and Victor's, p. 1371
Anatomy Recap: Why LMN Matters
The facial nerve (CN VII) has a long intratemporal course through the facial canal. Key landmarks:
- Greater petrosal nerve (preganglionic parasympathetics to lacrimal gland) branches off at the geniculate ganglion
- Nerve to stapedius - controls the stapedius muscle (sound dampening)
- Chorda tympani - taste to anterior 2/3 of tongue + parasympathetics to submandibular/sublingual glands
The level of the lesion in Bell's palsy is proximal to the chorda tympani in most cases, explaining taste loss and hyperacusis.
UMN vs. LMN facial palsy:
-
UMN (e.g., stroke): lower face only affected; forehead spared (bilateral cortical representation of frontalis)
-
LMN (Bell's palsy): entire ipsilateral face involved, including forehead and orbicularis oculi
-
Goldman-Cecil Medicine, p. 2962
Etiology and Pathophysiology
The cause is reactivation of latent Herpes Simplex Virus type 1 (HSV-1) in the geniculate ganglion - this is well-established by PCR evidence (Murakami et al., 1996, finding HSV-1 DNA in endoneurial fluid in 11/14 cases). The virus was also able to reproduce facial palsy when inoculated into mice.
- Varicella-zoster virus (VZV) is the second most frequent viral cause (up to one-third of cases)
- SARS-CoV-2 and HIV seroconversion have also been implicated
- Pathologically: mononuclear cell infiltration of the facial nerve, edema, swelling - the nerve becomes compressed within the tight bony facial canal
Risk factors:
-
Pregnancy (especially third trimester and first 2 weeks postpartum - up to 3-fold increased risk)
-
Diabetes mellitus
-
Hypertension (possible)
-
Recurrence in ~7-8% of cases; mean interval between episodes ~10 years
-
Harrison's, p. 3597 | Adams and Victor's, pp. 1371-1372
Clinical Features
Onset
- Abrupt - maximal weakness by 48 hours as a rule (practically all within 3-4 days)
- Patient often notices on inspection in the mirror in the morning
Symptoms and Signs
| Feature | Mechanism |
|---|---|
| Unilateral facial weakness (all divisions) | LMN CN VII palsy |
| Inability to close eye (lagophthalmos) | Orbicularis oculi paralysis |
| Bell's phenomenon | Upward rolling of the eye on attempted lid closure (protective reflex) |
| Drooping of corner of mouth, drooling | Orbicularis oris / buccinator paralysis |
| Loss of nasolabial fold | Ipsilateral |
| Pain behind the ear | Precedes palsy by 1-2 days (mastoid pain) |
| Loss of taste (anterior 2/3 tongue) | Chorda tympani involvement |
| Hyperacusis | Stapedius muscle paralysis |
| Mild facial numbness/fullness | (Uncommon; trigeminal overlap) |
| Mild CSF lymphocytosis | Occasional finding |
- Harrison's, p. 3597 | Adams and Victor's, p. 1371
MRI Findings
MRI with gadolinium (fat-suppressed T1) shows diffuse, smooth linear enhancement of the facial nerve - involving the geniculate ganglion, tympanic, and mastoid segments within the temporal bone - without a mass lesion.
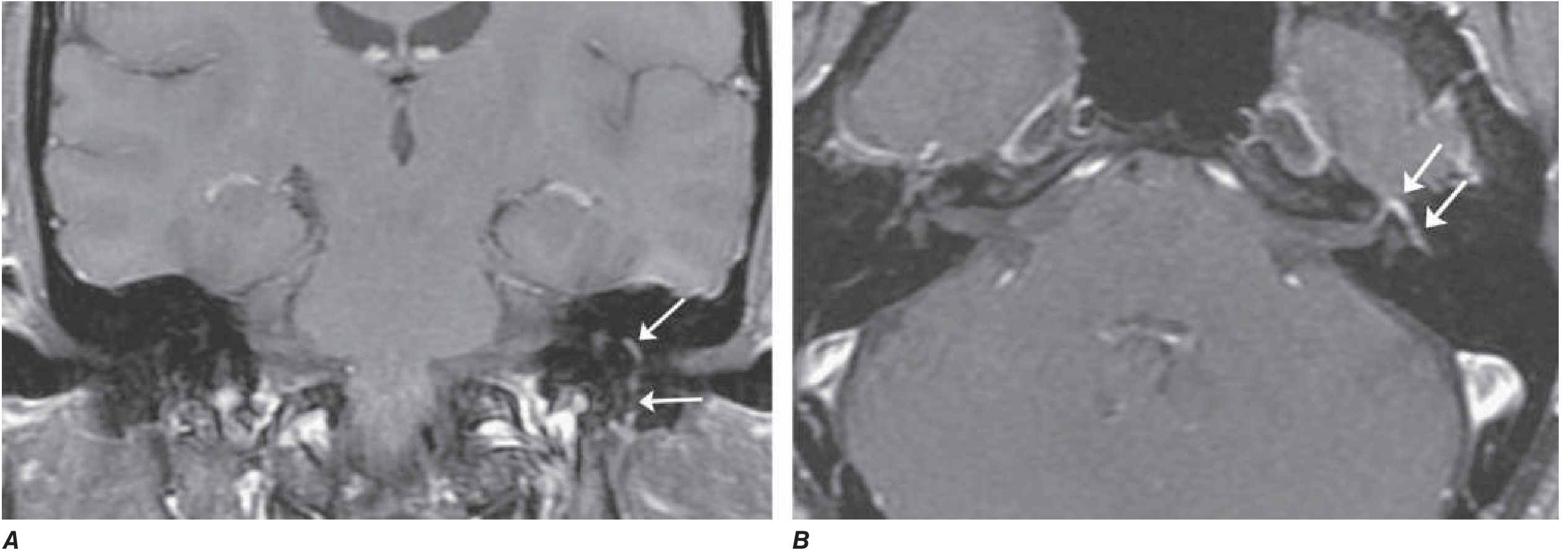
More pronounced enhancement correlates with a worse prognosis. - Harrison's, p. 3598 | Adams and Victor's, p. 1371
Differential Diagnosis
Bell's palsy is a diagnosis of exclusion. Always consider:
| Condition | Key distinguishing features |
|---|---|
| Ramsay Hunt Syndrome (VZV reactivation) | Vesicular rash in external auditory canal, pinna, palate; CN VIII involvement; more severe palsy; worse prognosis |
| Lyme disease | Endemic area; erythema migrans; can be bilateral; CSF pleocytosis; treat with antibiotics |
| Otitis media / mastoiditis | Abnormal otoscopy; hearing loss; fever |
| Parotid tumor | Slowly progressive; palpable mass; no taste loss |
| Facial nerve neuroma | Recurrent or slowly progressive palsy |
| Sarcoidosis | Often bilateral palsy; hilar lymphadenopathy; elevated ACE |
| Guillain-Barré syndrome | Bilateral facial palsy; ascending weakness; areflexia |
| HIV seroconversion | CSF pleocytosis |
| Carcinomatous meningitis | Multiple cranial nerve palsies; abnormal CSF cytology |
| Melkersson-Rosenthal syndrome | Recurrent facial palsy + facial edema + fissured tongue |
| Stroke / UMN lesion | Forehead spared; contralateral body weakness |
| Leprosy | Endemic area; skin lesions; sensory loss |
| Diabetes mellitus | Background; mononeuropathy |
- Harrison's, p. 3598 | Goldman-Cecil, p. 2962 | Adams and Victor's, p. 1372
Investigations
In straightforward Bell's palsy, investigations are not always required. However:
-
MRI - not routine; useful if atypical features, slow progression, or suspected central cause
-
Serology - consider Borrelia (Lyme), HIV, VZV in appropriate clinical context
-
EMG/nerve conduction - prognostic value:
- Evidence of denervation after 10 days = axonal degeneration = expect long recovery (months to years), potentially incomplete
- Early motor recovery (days 5-7) = most favorable prognostic sign
- Recovery of taste in first week = good prognostic sign
-
Harrison's, p. 3597 | Adams and Victor's, p. 1371
Treatment
1. Corticosteroids (First-line)
- Prednisolone 25 mg twice daily (50 mg/day) for 10 days, started early
- Evidence: Large RCTs (Sullivan et al., Engstrom et al.) show steroids increase return of facial function from 63% to 83% at 3 months
- Mechanism: reduces nerve swelling and edema within the bony facial canal
- Goldman-Cecil, p. 2973 | Adams and Victor's, p. 1372
2. Antiviral Agents
- Acyclovir alone - no independent benefit over placebo
- Valacyclovir + prednisolone - additive benefit shown in severe/complete palsy (Hato et al.) vs. prednisolone alone; particularly for complete facial palsy
- Acyclovir 400 mg 5x/day for 7 days; double dose for VZV-suspected cases
- Note: Ramsay Hunt syndrome should receive antivirals (valacyclovir/acyclovir) + steroids
- Goldman-Cecil, p. 2973 | Adams and Victor's, p. 1372
3. Eye Care (Essential)
- Corneal protection is mandatory - inability to close the eye leads to corneal exposure, dryness, and risk of ulceration
- Lubricating eye drops during the day; eye ointment + eye patch/tape at night
4. Physical Therapy
- Facial exercises may help maintain muscle tone and promote neuromuscular retraining
- A 2025 systematic review (Varelas et al., Am J Otolaryngol, 2025) supports a role for physical therapy in idiopathic facial paralysis
5. Surgical Decompression
- No evidence of benefit; may be harmful. Not recommended. - Adams and Victor's, p. 1372
Children
- Recovery rate is very high (~90%) regardless of treatment; steroid use is not mandatory in children - Scott-Brown's Otorhinolaryngology
Prognosis
- ~70-80% recover completely within 1-2 months
- ~85% achieve near-normal function overall
- ~10% have little or no recovery
- Unfavorable signs: complete palsy, denervation on EMG after 10 days
- Favorable signs: incomplete palsy in first week, early taste recovery, early motor recovery (days 5-7)
Complications of Incomplete Recovery
-
Synkinesis - aberrant nerve regeneration causing involuntary co-contractions:
- "Jaw winking" (eye closes when mouth opens)
- Crocodile tears (lacrimation accompanying salivation) - from aberrant regeneration of parasympathetic fibers
-
Facial contracture
-
Hemifacial spasm (rare, late)
-
Goldman-Cecil, p. 2978 | Adams and Victor's, p. 1372
Summary Table
| Feature | Detail |
|---|---|
| Incidence | ~25/100,000/year; 1 in 60 lifetime |
| Cause | HSV-1 reactivation in geniculate ganglion; VZV 2nd most common |
| Onset | Abrupt; max weakness by 48h |
| Pattern | Unilateral LMN (all face including forehead) |
| Key symptoms | Facial weakness, lagophthalmos, post-auricular pain, taste loss, hyperacusis |
| MRI | Linear gadolinium enhancement of facial nerve in temporal bone |
| Treatment | Prednisolone 50 mg/day x10 days ± valacyclovir; eye care |
| Prognosis | 85% near-complete recovery; ~10% poor outcome |
| Recurrence | ~7-8% |
Dementia
Finding Sources
Searching PubMed
"dementia"[MeSH Terms] AND "diagnosis OR treatment"
Finding Sources
Searching PubMed
"Alzheimer disease"[MeSH Terms] AND "therapeutics"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
I now have comprehensive information from Harrison's 22E (2025) covering definition, functional anatomy, causes, differential diagnosis, and treatment. Let me compile the full answer now.
Dementia
Definition
Dementia is an acquired, persistent deterioration in cognitive abilities that impairs the successful performance of activities of daily living (ADLs). It affects over 6 million people in the United States, with a total annual healthcare cost exceeding $300 billion. Prevalence rises sharply with age: 10% of persons >70 years and 20-40% of individuals >85 years have clinically identifiable memory loss.
Key points:
-
Episodic memory (recall of time/place-specific events) is the most commonly lost cognitive function
-
Other domains affected: language, visuospatial function, praxis, calculation, judgment, executive function
-
Neuropsychiatric symptoms are common: depression, apathy, anxiety, hallucinations, delusions, agitation, disinhibition
-
A preclinical stage (brain pathology present, no symptoms) and a prodromal stage - Mild Cognitive Impairment (MCI) (cognitive decline but ADLs preserved) precede overt dementia in neurodegenerative disease
-
Harrison's 22E, p. (Ch. 31)
Functional Anatomy
Different dementia syndromes begin in distinct brain regions:
| Region Initially Affected | Dementia Type | Early Presentation |
|---|---|---|
| Entorhinal cortex → hippocampus → limbic → parietal cortex | Alzheimer's disease (AD) | Episodic memory loss |
| Frontal/prefrontal cortex, anterior temporal | Frontotemporal dementia (FTD) | Behavioral change, language, executive dysfunction |
| Cortical + subcortical (patchwork) | Vascular dementia | Executive dysfunction, psychomotor slowing |
| Posterior cortical (parietal/occipital) | Dementia with Lewy bodies (DLB) | Visuospatial, hallucinations |
| Subcortical (striatum, basal ganglia) | Huntington's disease | Movement, executive, mood |
Ascending cholinergic pathways are critical for attention and memory (hence cholinesterase inhibitors as therapy). Noradrenergic, serotonergic, and dopaminergic pathways modulate behavior and mood. - Harrison's 22E
Causes of Dementia
A. Neurodegenerative (Progressive, Irreversible)
| Disease | Key Pathology | Key Features |
|---|---|---|
| Alzheimer's disease (AD) | Amyloid-β plaques + tau neurofibrillary tangles | Most common (~60-70%); episodic memory first |
| Dementia with Lewy bodies (DLB) | α-synuclein (Lewy bodies) in cortex | Visual hallucinations, parkinsonism, REM sleep behavior disorder, cognitive fluctuation |
| Frontotemporal Dementia (FTD) | Tau or TDP-43 aggregates | Behavioral disinhibition/apathy OR progressive aphasia; spares memory early |
| Parkinson's disease dementia (PDD) | α-synuclein (subcortical) | Motor Parkinson's precedes dementia by ≥1 year |
| LATE (Limbic-predominant Age-related TDP-43 Encephalopathy) | TDP-43 aggregates | Mimics AD; increasingly common >90 years; >50% of dementia at age >90 |
| Huntington's disease | CAG repeat expansion in HTT gene | Chorea, executive dysfunction, psychiatric |
| Progressive supranuclear palsy (PSP) | Tau; subcortical | Vertical gaze palsy, axial rigidity, early falls |
| Prion disease (CJD) | Misfolded PrP | Rapid progression; myoclonus; MRI cortical ribboning |
B. Vascular
- Cortical/subcortical infarctions (multi-infarct dementia)
- Confluent white matter disease (leukoaraiosis), CADASIL
- Small vessel disease
C. Reversible / Treatable Causes (Critical to identify)
| Cause | Treatment |
|---|---|
| Hypothyroidism | Thyroid replacement |
| Vitamin B12 deficiency | Parenteral B12 |
| Thiamine (B1) deficiency (Wernicke's) | IV thiamine |
| Neurosyphilis | Penicillin |
| HIV-associated dementia | Antiretrovirals |
| Normal pressure hydrocephalus (NPH) | Ventriculoperitoneal shunting |
| CNS neoplasm | Surgery, radiation, chemotherapy |
| Chronic subdural hematoma | Surgical drainage |
| Drug/medication toxicity | Remove offending agent |
| Depression ("pseudodementia") | Antidepressants |
| Hypercalcemia, hyponatremia | Correct metabolic disturbance |
| Chronic CNS infections (TB meningitis, fungal) | Antimicrobials |
| Autoimmune/paraneoplastic encephalitis (anti-NMDAR, LGI1, etc.) | Immunotherapy |
- Harrison's 22E
Differential Diagnosis: Key Features Side-by-Side
| Disease | First Symptom | Mental Status | Neuropsychiatry | Neurology | Imaging |
|---|---|---|---|---|---|
| AD | Memory loss | Episodic memory loss; executive, language, visuospatial | Irritability, anxiety, depression | Initially normal | Entorhinal/hippocampal atrophy; posterior-predominant |
| Vascular | Often sudden; variable; apathy, falls | Frontal/executive; cognitive slowing; can spare memory | Apathy, delusions, anxiety | Motor slowing, spasticity | Cortical/subcortical infarctions; white matter disease |
| DLB | Visual hallucinations, REM sleep behavior disorder, Capgras syndrome, parkinsonism | Drawing/frontal-executive; spares memory; delirium-prone | Visual hallucinations, depression, sleep disorder | Parkinsonism | Posterior parietal atrophy; hippocampi larger than AD |
| LATE | Memory loss | Episodic memory; mild semantic deficits | None described | Normal | Medial temporal/hippocampal atrophy (anterior predominant) |
| FTD | Apathy, poor judgment/insight, speech/language, hyperorality | Frontal/executive and/or language; spares drawing | Apathy, disinhibition, overeating, compulsivity | May have vertical gaze palsy, MND | Frontal, insular, anterior temporal atrophy; spares posterior parietal |
| CJD | Dementia, mood, anxiety, movement | Variable; frontal/executive; focal cortical | Depression, anxiety, psychosis | Myoclonus, rigidity, parkinsonism | Cortical ribboning + basal ganglia/thalamic hyperintensity on DWI/FLAIR |
From Harrison's 22E, Table 31 (Ch. 31)
Alzheimer's Disease - Key Details
Epidemiology: Most common dementia (~60-70%). Most are sporadic late-onset (age >65). Early-onset familial AD (<65 years) accounts for ~5%.
Genetics:
- APOE ε4 allele: major genetic risk factor for late-onset sporadic AD
- APP, PSEN1, PSEN2 mutations: autosomal dominant early-onset AD
- Down syndrome (Trisomy 21): virtually all develop AD pathology by age 40-50 (APP gene on chromosome 21)
Pathology (amyloid cascade hypothesis):
- Senile plaques: extracellular deposits of amyloid-β (Aβ) peptide (from cleavage of APP)
- Neurofibrillary tangles (NFTs): intracellular aggregates of hyperphosphorylated tau protein
- Neuronal loss and synapse loss - particularly cholinergic neurons of the nucleus basalis of Meynert
- Spreads in a predictable Braak staging pattern: entorhinal cortex → hippocampus → association cortex → diffuse
Biomarkers (amyloid-tau-neurodegeneration [ATN] framework):
- CSF: low Aβ42, elevated total tau and phospho-tau
- Amyloid PET: positive for Aβ plaques
- Tau PET: elevated tau tangles
- MRI: hippocampal and entorhinal atrophy, posterior cortical predominance
- FDG-PET: hypometabolism in temporal-parietal regions
Frontotemporal Dementia - Key Details
FTD encompasses three core clinical syndromes:
- Behavioral variant (bvFTD) - most common; apathy, disinhibition, compulsivity, loss of empathy, hyperorality
- Semantic variant PPA - loss of word/object/person meaning; fluent but empty speech
- Nonfluent/agrammatic PPA - effortful, halting speech; motor speech impairment
Genetics: Autosomal dominant in 10-20%; mutations in C9orf72 (hexanucleotide GGGGCC expansion), GRN (granulin), MAPT (tau) on chromosome 17.
FTD-MND (with motor neuron disease) is a serious overlap syndrome. - Harrison's 22E
Dementia with Lewy Bodies - Key Features
Core clinical features:
- Fluctuating cognition (variable attention and alertness)
- Recurrent visual hallucinations (well-formed, detailed)
- REM sleep behavior disorder (RBD) - often precedes dementia by years
- Parkinsonism (bradykinesia, rigidity, tremor)
Important caveat: DLB patients are exquisitely sensitive to antipsychotics (neuroleptic sensitivity) - typical antipsychotics can cause irreversible worsening or death. Avoid haloperidol and other D2 blockers. Memantine also used with great caution. - Harrison's 22E
Diagnosis
History: Onset, progression (insidious vs. stepwise), first domain affected, neuropsychiatric features, family history, medications, vascular risk factors
Cognitive testing: MMSE, MoCA (better for MCI), MOCA-Blind; formal neuropsychological testing
Investigations:
- Essential bloods: TSH, B12, folate, CBC, CMP, LFTs, glucose, calcium, VDRL/RPR, HIV in appropriate cases
- Brain MRI (preferred over CT) - structural atrophy pattern, vascular disease, NPH
- Lumbar puncture: if CNS infection, inflammatory/autoimmune dementia, or paraneoplastic suspected; CSF biomarkers (Aβ42, tau) for AD
- EEG: useful in CJD (periodic sharp wave complexes), and to exclude seizure-related cognitive decline
- PET: FDG-PET (hypometabolism pattern), amyloid PET, tau PET - increasingly used to confirm AD diagnosis
- Genetic testing: APOE genotyping (risk), familial AD gene panel in early-onset cases
Treatment
1. Treat Reversible Causes
See the reversible causes table above - always exclude before accepting a neurodegenerative diagnosis.
2. Cholinesterase Inhibitors (AChEIs)
- Mechanism: inhibit acetylcholinesterase → increase synaptic acetylcholine
- Agents: Donepezil (AD), Rivastigmine (AD, PDD), Galantamine (AD)
- Modest symptomatic benefit in mild-moderate AD and PDD
- Side effects: GI (nausea, diarrhea), bradycardia, vivid dreams
3. Memantine
- Mechanism: NMDA glutamate receptor antagonist; reduces excitotoxic neuronal damage
- Indicated for moderate to severe AD
- Combination with a cholinesterase inhibitor delayed nursing home placement in some studies
- Use with great caution in DLB (risk of worsening agitation)
4. Anti-Amyloid Monoclonal Antibodies (new, disease-modifying)
- Lecanemab (Leqembi) - FDA approved for early AD (MCI/mild dementia with confirmed amyloid); reduces amyloid burden and slows clinical decline (van Dyck et al., NEJM 2023 [PMID referenced in Harrison's])
- Donanemab - FDA approved 2024; similar mechanism; significant slowing of decline in TRAILBLAZER-ALZ 2 trial (Sims et al., JAMA 2023)
- Key risk: ARIA (Amyloid-Related Imaging Abnormalities) - brain edema/microhemorrhages on MRI; APOE ε4 homozygotes at highest risk
5. Behavioral/Neuropsychiatric Symptoms
- First: identify and correct reversible triggers (pain, infection, constipation, UTI, electrolyte disturbance, drug toxicity, hunger)
- Depression: SSRIs (escitalopram 5-10 mg/day starting) or SNRIs - few cognitive side effects; preferred
- Agitation/aggression/psychosis: second-generation antipsychotics (e.g., quetiapine 12.5-25 mg starting) - significant risk including increased mortality in elderly with dementia; use cautiously; brexpiprazole is now first FDA-approved drug specifically for AD-related agitation
- Apathy, visual hallucinations in DLB: cholinesterase inhibitors often help, avoiding need for antipsychotics
- Avoid in DLB: typical antipsychotics, memantine (high doses), anticholinergics, benzodiazepines
6. Anticonvulsants
- Levetiracetam or lamotrigine preferred if seizures develop (favorable cognitive profile, and animal model evidence of possible cognitive benefit in AD)
7. Non-Pharmacological
-
Cognitive stimulation, physical exercise, sleep hygiene
-
Mediterranean diet - associated with reduced risk of cognitive impairment and AD in a 2025 meta-analysis (Fekete et al., Geroscience, 2025)
-
Caregiver education and support; advance care planning
-
Harrison's 22E (Ch. 31 + Ch. 442)
Prognosis
| Dementia Type | Course | Mean Survival from Diagnosis |
|---|---|---|
| AD | Slow progressive (8-10 years average) | 8-10 years |
| DLB | Progressive with fluctuations | Shorter than AD; ~5-7 years |
| FTD | Variable; 6-8 years | 6-8 years; shorter with MND overlap |
| Vascular | Stepwise or gradual | Depends on vascular events |
| CJD | Rapid (months) | <1 year from symptom onset |
| NPH | Reversible with shunting | Good if treated early |
Late-onset dementia is rarely "pure" pathology - at autopsy, most patients with dementia have 3-4 concurrent pathologies. In one large cohort (mean age at death 89.7 years), AD neuropathology explained ~39% of attributable dementia risk, vascular pathology ~25%, with the rest from LATE, DLB, and mixed pathology. - Harrison's 22E
Summary: DEMENTIA Mnemonic for Reversible Causes
| Letter | Cause |
|---|---|
| D | Drugs/Deficiencies (B12, thiamine, folate) |
| E | Endocrine (hypothyroidism, Cushing's, hypercalcemia) |
| M | Metabolic (hyponatremia, hepatic/renal failure) |
| E | Emotional/psychiatric (depression = pseudodementia) |
| N | Neoplasm (CNS tumor, paraneoplastic) |
| T | Trauma (chronic subdural hematoma) |
| I | Infections (neurosyphilis, HIV, TB meningitis, viral encephalitis) |
| A | Autoimmune/vascular (anti-NMDAR encephalitis, NPH, vasculitis) |
Ataxia
Finding Sources
Searching PubMed
"cerebellar ataxia"[MeSH Terms] AND "diagnosis"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive information. Let me compile the full answer.
Ataxia
Definition
Ataxia refers to a disturbance in the smooth, coordinated performance of voluntary motor acts - causing incoordination or impaired balance. Movements are flawed in rate, range, timing, direction, and force. In the absence of cerebellar inhibitory and modulating influences, skilled movements become inaccurate and poorly controlled. Ataxia may affect the limbs, trunk, or gait. - Localization in Clinical Neurology, 8e
Types of Ataxia
There are three main types based on the underlying mechanism:
1. Cerebellar Ataxia
Caused by dysfunction of the cerebellum or its connections (spinocerebellar tracts, cerebellar peduncles).
Clinical features:
- Wide-based, lurching, irregular gait
- Dysmetria - overshooting/undershooting targets (finger-nose, heel-shin tests abnormal)
- Dysdiadochokinesia - inability to perform rapid alternating movements
- Intention tremor - tremor that worsens on approaching a target
- Nystagmus - often horizontal or gaze-evoked
- Dysarthria - scanning (staccato) speech
- Truncal titubation - rhythmic oscillation of the trunk/head
- Hypotonia - decreased muscle tone ipsilateral to cerebellar lesion
- Rebound phenomenon (failure of check)
- Romberg test: may be positive but less dramatically than sensory ataxia
2. Sensory Ataxia
Due to loss of proprioception (large fibre afferents) from peripheral nerves, dorsal root ganglia, or posterior columns of spinal cord.
Clinical features:
- Wide-based gait; patient looks down at feet when walking ("stamping gait")
- Feet thrust forward with slapping sound ("slapping gait")
- Worsens dramatically in the dark and on uneven surfaces
- Romberg sign positive - falls when eyes closed (critically distinguishes from cerebellar ataxia)
- Loss of joint position sense and vibration in lower limbs
- Heel-shin test: relatively preserved compared to cerebellar
3. Vestibular Ataxia
Due to vestibular organ or pathway dysfunction.
Clinical features:
- Veering/leaning toward the side of the lesion
- Severe vertigo, nausea, nystagmus
- Unsteadiness unmasked during head rotation while walking
- Less severe when running vs. walking (in acute vestibular disease)
Comparison Table: The Three Ataxias
| Feature | Cerebellar Ataxia | Sensory Ataxia | Frontal Gait Disorder |
|---|---|---|---|
| Base of support | Wide | Wide; looks down | Wide |
| Velocity | Variable | Slow | Very slow |
| Stride | Irregular, lurching | Regular with path deviation | Short, shuffling |
| Romberg test | +/- | Unsteady, falls (positive) | +/- |
| Heel-shin test | Abnormal | +/- | Normal |
| Initiation | Normal | Normal | Hesitant |
| Turns | Unsteady | +/- | Hesitant, multistep |
| Falls | Late event | Frequent | Frequent |
From Harrison's 22E, Table 26-3
Cerebellar Anatomy and Localization
| Cerebellar Region | Function | Lesion Effect |
|---|---|---|
| Vermis (midline) | Truncal/axial coordination, gait | Truncal ataxia, gait ataxia, titubation |
| Hemispheres (lateral) | Limb coordination (ipsilateral) | Limb ataxia, dysmetria, dysdiadochokinesia |
| Flocculonodular lobe | Vestibular-ocular coordination | Nystagmus, vertigo, gait imbalance |
| Deep nuclei (dentate) | Output to thalamus/cortex | Intention tremor |
Causes of Ataxia by Time Course
Acute Ataxia (Hours to Days)
| Category | Examples |
|---|---|
| Toxic/Drug | Alcohol intoxication, phenytoin, lithium, barbiturates, carbamazepine |
| Metabolic | Hypoglycemia, hyponatremia, hyperammonemia, Wernicke's encephalopathy (thiamine B1 deficiency) |
| Vascular | Cerebellar infarction, cerebellar hemorrhage |
| Infectious | Bacterial/viral meningitis; acute postinfectious cerebellar ataxia (varicella - especially in children) |
| Immune | Fisher variant of GBS (anti-GQ1b; ataxia + ophthalmoplegia + areflexia), Bickerstaff brainstem encephalitis |
| Biotinidase deficiency | Treatable metabolic disease |
| Trauma | Posterior fossa hemorrhage |
Subacute Ataxia (Weeks to Months)
| Category | Examples |
|---|---|
| Paraneoplastic | Anti-Yo (breast, ovarian Ca), anti-Hu (small-cell lung Ca), anti-Tr (Hodgkin's) |
| Autoimmune | Anti-GAD65 antibodies (most common autoimmune cerebellar ataxia); anti-gliadin/anti-tissue transglutaminase (gluten ataxia) |
| Alcohol + malnutrition | Cerebellar vermis degeneration (B vitamins deficiency) |
| Prion disease | CJD - rapidly progressive ataxia with myoclonus, dementia |
| Toxic | Heavy metals (mercury), toluene (glue/solvent sniffing), chemotherapy (5-FU, paclitaxel) |
Chronic Progressive Ataxia (Months to Years)
| Category | Examples |
|---|---|
| Hereditary | SCAs (autosomal dominant), Friedreich's ataxia (AR), ataxia-telangiectasia |
| Multiple system atrophy - cerebellar (MSA-C) | Progressive with autonomic failure |
| Hypothyroidism | Reversible; always check TFTs |
| Vascular | Chronic white matter disease, CADASIL |
| Paraneoplastic | Slow-growing tumor, long latency |
| Infectious | Meningovascular syphilis, tabes dorsalis (posterior column degeneration) |
Inherited Ataxias
Autosomal Dominant: Spinocerebellar Ataxias (SCAs)
SCAs include SCA1 through SCA50+, caused by mutations in different genes. The most common are:
| SCA Type | Gene/Mechanism | Distinguishing Features |
|---|---|---|
| SCA1 | CAG repeat ↑ in ATXN1 (chr 6) | Progressive cerebellar ataxia, nystagmus, dysarthria, pyramidal signs, extrapyramidal features |
| SCA2 | CAG repeat ↑ in ATXN2 (chr 12) | Slow saccades (very characteristic), hyporeflexia, peripheral neuropathy |
| SCA3 (Machado-Joseph disease) | CAG repeat ↑ in ATXN3 (chr 14) - most common SCA worldwide | Ataxia + parkinsonism/dystonia/pyramidal signs; "bulging eyes" (eyelid retraction) |
| SCA6 | CAG repeat ↑ in CACNA1A (chr 19) - calcium channel | Pure cerebellar ataxia; late onset; relatively mild |
| SCA7 | CAG repeat ↑ in ATXN7 | Ataxia + progressive retinal degeneration (pigmentary macular dystrophy → blindness) |
| SCA17 | CAG repeat ↑ in TBP (TATA-binding protein) | Ataxia + dementia + psychiatric symptoms |
Mechanism common to CAG-repeat SCAs: CAG encodes glutamine → expanded polyglutamine proteins (ataxins) → toxic gain of function, protein aggregation, neuronal apoptosis. Anticipation occurs (earlier onset in successive generations with increasing CAG repeat number).
Episodic Ataxia (EA):
- EA-1: KCNA1 potassium channel mutation (chr 12p); brief episodes (minutes); myokymia between episodes; triggered by startle/posture change
- EA-2: CACNA1A calcium channel mutation (chr 19p); longer episodes (hours-days); gaze-evoked nystagmus between episodes; stress/exercise triggers; treat with acetazolamide
DRPLA (Dentatorubropallidoluysian Atrophy): CAG repeat in ATROPHIN gene (chr 12p); ataxia + chorea + myoclonus + seizures + dementia; common in Japan
Autosomal Recessive Ataxias
Friedreich's Ataxia - Most Common Inherited Ataxia (~50% of all hereditary ataxias)
Genetics: GAA trinucleotide repeat expansion in intron 1 of the frataxin (FXN) gene on chromosome 9q → deficiency of frataxin protein → mitochondrial iron accumulation → oxidative stress → neurodegeneration
Clinical features (onset <25 years - most present in adolescence):
- Progressive gait ataxia and frequent falls; lower limbs more affected than upper
- Dysarthria (scanning speech)
- Absent deep tendon reflexes (areflexia) - lost early
- Extensor plantar responses (Babinski sign) - UMN involvement
- Loss of vibration and proprioception (posterior column degeneration)
- Nystagmus, loss of fast saccades
- Pes cavus (high-arched feet) and scoliosis - characteristic deformities
- Cardiomyopathy (90%) - hypertrophic cardiomyopathy, conduction defects; leading cause of death
- Diabetes mellitus (20%)
- Median age of death: 35 years; women have better prognosis than men
Pathology: Degeneration of spinocerebellar tracts, posterior columns, lateral corticospinal tracts, dorsal root ganglia; slight cerebellar/cerebral atrophy
Treatment: Omaveloxolone (Skyclarys) - FDA-approved (2023) for Friedreich's ataxia in patients ≥16 years; activates Nrf2 pathway, reduces oxidative damage; slows neurological progression. Symptomatic: physical therapy, cardiac management, orthopedic care for scoliosis/foot deformities.
Ataxia-Telangiectasia (AT)
- Gene: ATM (chr 11q22-23) - DNA repair kinase; autosomal recessive
- Features: Progressive cerebellar ataxia from infancy, oculocutaneous telangiectasias (conjunctiva, ears, skin), immune deficiency (IgA, IgE deficiency; T-cell dysfunction), increased cancer risk (lymphomas, leukemias), sinopulmonary infections, elevated AFP
- Radiosensitivity - avoid radiation exposure (increased DNA damage); cancer risk with radiotherapy
Investigations
| Test | Indication |
|---|---|
| MRI brain + spine | Structural cause, cerebellar/spinal atrophy pattern; infarction, hemorrhage, tumor |
| TFTs (TSH) | Hypothyroid ataxia (reversible) |
| Vitamin B12, B1 (thiamine) | Nutritional/malabsorptive |
| Glucose, electrolytes (Na, Ca) | Metabolic ataxia |
| Toxicology screen | Drug/alcohol intoxication |
| Anti-gliadin, tTG antibodies | Gluten ataxia |
| Paraneoplastic panel | Anti-Yo, Anti-Hu, Anti-Tr, anti-GAD65 |
| CSF analysis | Prion disease (14-3-3 protein, RT-QuIC), immune-mediated ataxia |
| Genetic testing | SCA panel (CAG repeat sizing), Friedreich's (GAA repeat), AT (ATM mutation) |
| EMG/NCS | Peripheral neuropathy (sensory ataxia), Friedreich's (absent sensory potentials) |
| Echocardiogram | Friedreich's ataxia (cardiomyopathy screening) |
| VDRL/RPR | Tabes dorsalis |
| Karyotype | If clinical features of chromosomal disease |
| FDG-PET / DAT scan | MSA-C vs. Parkinson-plus |
Treatment
Specific/Curative
| Cause | Treatment |
|---|---|
| Episodic ataxia EA-1/EA-2 | Acetazolamide (EA-2 especially); anticonvulsants (EA-1) |
| Hypothyroidism | Thyroid hormone replacement |
| Gluten ataxia | Strict gluten-free diet |
| Vitamin deficiency | Thiamine (Wernicke's), B12, vitamin E |
| Autoimmune/paraneoplastic | IV methylprednisolone, IVIG, plasma exchange; treat underlying cancer |
| Friedreich's ataxia | Omaveloxolone (FDA-approved 2023); antioxidants; cardiac management |
| Drug toxicity | Remove offending agent |
| Cerebellar hemorrhage/infarct | Surgical decompression if mass effect; stroke risk factor management |
Symptomatic/Supportive
- Physiotherapy and occupational therapy - gait rehabilitation, walking aids, fall prevention
- Non-invasive brain stimulation (rTMS/tDCS) - emerging evidence; recent meta-analyses (Matsugi et al., Mov Disord Clin Pract, 2024) suggest modest benefit for degenerative cerebellar ataxia
- Speech therapy for dysarthria and dysphagia
- Baclofen or clonazepam for tremor
- Scoliosis/foot deformity - orthotics, surgical correction in Friedreich's ataxia
- Genetic counselling for hereditary ataxias
Summary: Approach to a Patient with Ataxia
Ataxia
├── Acute (<days) → Toxic (alcohol, drugs), metabolic, stroke, Fisher GBS
├── Subacute (weeks) → Paraneoplastic, autoimmune (anti-GAD65, gluten), Wernicke's
├── Chronic/hereditary → SCA (AD), Friedreich's (AR), MSA-C, AT
│
├── Cerebellar signs dominant → Cerebellum or its pathways
├── Sensory loss + positive Romberg → Posterior columns / large-fibre neuropathy
└── Vestibular features → Labyrinth / CN VIII / brainstem
Always exclude reversible causes first: hypothyroidism, vitamin deficiencies, alcohol/drug toxicity, gluten ataxia, and autoimmune/paraneoplastic disease before diagnosing a hereditary or degenerative condition.
Epilepsy
Finding Sources
Searching PubMed
"epilepsy"[MeSH Terms] AND "anticonvulsants"[MeSH Terms]
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive, detailed information from Harrison's 22E on epilepsy classification, causes, investigations, and treatment. Let me compile the full answer.
Epilepsy
Definition
Epilepsy is defined as two or more unprovoked seizures occurring more than 24 hours apart. A seizure is a paroxysmal event caused by abnormal, excessive, hypersynchronous neuronal discharges from the cerebral cortex.
- Incidence: ~0.3-0.5% of the population worldwide
- Prevalence: 5-30 persons per 1000
- About 30% of patients fail to respond to standard antiseizure medications (drug-resistant epilepsy)
Classification of Seizures (ILAE 2017)
The International League Against Epilepsy (ILAE) updated classification in 2017 divides seizures into three main categories:
1. Focal Onset Seizures
Originate within networks limited to one cerebral hemisphere (the older term "partial seizures" is no longer used). Further classified by:
- Awareness: intact (formerly "simple partial") or impaired (formerly "complex partial")
- Onset type: motor or nonmotor
- Evolution: may progress to focal to bilateral tonic-clonic seizures (formerly "secondary generalization")
Common focal seizure types by region of onset:
| Lobe | Typical Semiology |
|---|---|
| Temporal (most common) | Aura (déjà vu, rising epigastric sensation, fear), oral/manual automatisms, impaired awareness, postictal confusion |
| Frontal | Motor activity (tonic, clonic, hypermotor), brief, nocturnal, often no postictal state |
| Parietal | Contralateral sensory aura (tingling, numbness), body image distortion |
| Occipital | Visual aura (flashing lights, colored patterns), eye deviation, ictal blindness |
2. Generalized Onset Seizures
Arise within and rapidly engage networks distributed across both cerebral hemispheres.
| Type | Description |
|---|---|
| Tonic-clonic (GTC) | Tonic phase (stiffening, apnoea, cyanosis) → clonic phase (rhythmic jerking) → postictal drowsiness/confusion |
| Absence (typical) | Brief (5-30 sec) staring spell, sudden onset and offset, no postictal state; 3 Hz spike-wave on EEG |
| Absence (atypical) | Slower onset/offset; associated with cognitive impairment; part of Lennox-Gastaut syndrome |
| Myoclonic | Sudden, brief (<100 ms) muscle jerks, often bilateral; consciousness usually preserved |
| Tonic | Sustained muscle contraction; often nocturnal; risk of falls |
| Atonic ("drop attacks") | Sudden loss of muscle tone; fall to ground; risk of injury |
| Clonic | Rhythmic jerking without preceding tonic phase |
3. Unknown Onset
When the onset cannot be determined.
- Harrison's 22E, Ch. 436 (ILAE 2017 classification)
Causes of Epilepsy
The ILAE 2017 also classifies epilepsy by etiology:
Structural
- Hippocampal sclerosis (mesial temporal lobe epilepsy - most common form in adults)
- Cortical dysplasia / malformations of cortical development
- Tumors (primary or metastatic)
- Prior stroke/vascular malformation (AVM, cavernoma)
- Traumatic brain injury
- Periventricular leukomalacia (perinatal injury)
Genetic
- Ion channel mutations: SCN1A (Dravet syndrome), KCNQ2, CACNA1A
- Trinucleotide repeat disorders: fragile X, Huntington's (rarely)
- Chromosomal: Down syndrome, Angelman syndrome, ring chromosome 20
- Neurocutaneous syndromes: tuberous sclerosis (TSC1/TSC2), neurofibromatosis, Sturge-Weber
Infectious
- Neurocysticercosis (most common worldwide cause of acquired epilepsy)
- HSV encephalitis, HIV, tuberculoma, cerebral abscess
- Cerebral malaria
Metabolic
- Hypoglycemia, hyponatremia, hypocalcemia, hypomagnesemia
- Pyridoxine (B6) deficiency (neonates)
- Uraemia, hepatic encephalopathy
- Mitochondrial disorders
Immune / Autoimmune
- Anti-NMDAR encephalitis (most common autoimmune encephalitis)
- LGI1, CASPR2, GABA-B, AMPA receptor antibodies
- Rasmussen encephalitis
Drugs that Lower Seizure Threshold
- Bupropion, clozapine, lithium
- Beta-lactam antibiotics, quinolones, isoniazid
- Cocaine, amphetamine, phencyclidine
- Alcohol/benzodiazepine/barbiturate withdrawal
- Tramadol, meperidine, fentanyl
- Antimalarials (chloroquine, mefloquine)
- Cyclosporine, tacrolimus
Unknown (Idiopathic)
Large proportion, especially in children with genetic epilepsy syndromes.
Epilepsy Syndromes
Key recognizable syndromes by age of onset:
| Syndrome | Age of Onset | Seizure Types | EEG | Outcome |
|---|---|---|---|---|
| Benign neonatal seizures | Neonatal | Focal clonic | Variable | Good |
| West syndrome (infantile spasms) | 3-12 months | Epileptic spasms + developmental regression | Hypsarrhythmia | Poor if untreated |
| Dravet syndrome | <1 year | Febrile + afebrile; drug-resistant | Abnormal | Poor; SCN1A mutation |
| Lennox-Gastaut syndrome | 1-7 years | Multiple types (tonic, atonic, atypical absence) | Slow spike-wave (<2.5 Hz) | Poor; intellectual disability |
| Childhood absence epilepsy | 4-10 years | Typical absence | 3 Hz spike-wave | Good; often remits |
| Juvenile absence epilepsy | Adolescence | Absence + occasional GTC | 3-4 Hz spike-wave | Moderate; lifelong in many |
| Juvenile myoclonic epilepsy (JME) | 12-18 years | Morning myoclonic jerks + GTC + absence | 3.5-4 Hz polyspike-wave | Good with medication; but lifelong |
| Mesial temporal lobe epilepsy (MTLE) | Any (often adolescence) | Focal with impaired awareness; oral automatisms | Temporal sharp waves | Often drug-resistant; surgical candidate |
JME is particularly important: seizures triggered by sleep deprivation and alcohol; EEG shows bilateral polyspike-wave at 3.5-4 Hz; responds to valproate; requires lifelong treatment.
Pathophysiology
Two key mechanisms of seizure generation:
- Increased excitation: enhanced glutamatergic transmission (NMDA, AMPA receptors); mutation in Na+/Ca2+ channels causing increased neuronal firing
- Decreased inhibition: reduced GABA-ergic inhibition (GABA-A receptor dysfunction); loss of inhibitory interneurons
The ictal discharge begins with paroxysmal depolarization shifts (PDS) in neurons - large depolarizations driven by NMDA receptor activation - causing a burst of action potentials. Hypersynchronisation of neuronal populations within a focus then spreads.
Diagnosis
History
- Detailed seizure description from patient AND witness (onset, duration, postictal state, aura)
- Risk factors: prior febrile seizures, family history, head trauma, stroke, CNS infection
- Precipitating factors: sleep deprivation, alcohol, drugs
- Developmental history (children)
Investigations
| Test | Purpose |
|---|---|
| EEG | Primary test; epileptiform discharges in ~90% of epilepsy; 3 Hz spike-wave in absence; temporal sharp waves in TLE; normal EEG does not exclude epilepsy |
| MRI brain | Structural cause; hippocampal sclerosis, dysplasia, tumor, AVM; preferred over CT |
| Blood tests | Glucose, electrolytes (Na, Ca, Mg), LFTs, urea, FBC; toxicology screen |
| Lumbar puncture | Suspected CNS infection/encephalitis; mandatory in HIV |
| Autoantibody panel | Anti-NMDAR, LGI1, CASPR2, GABA-B - serum and CSF |
| Genetic testing | Suspected genetic epilepsy; SCN1A (Dravet), KCNQ2, chromosomal microarray |
| Video-EEG monitoring | Gold standard for seizure classification and pre-surgical evaluation |
| FDG-PET / SPECT | Pre-surgical evaluation for drug-resistant focal epilepsy (interictal hypometabolism / ictal hyperperfusion) |
| Neuropsychological testing | Cognitive profiling; lateralisation pre-surgery |
EEG pearls:
- Only ~2% of people without epilepsy have epileptiform discharges
- ~90% of epilepsy patients show epileptiform activity, depending on recording circumstances
- 3 Hz spike-wave: absence epilepsy
- Hypsarrhythmia: West syndrome (infantile spasms)
- Periodic sharp wave complexes: CJD
- Normal EEG does not exclude epilepsy (especially temporal lobe or frontal lobe)
Antiseizure Drug Therapy
When to Start
- After two or more unprovoked seizures
- After one unprovoked seizure if high risk of recurrence (structural lesion, abnormal EEG, nocturnal seizure, neurological deficit)
Principles
- Monotherapy is the goal; start low, go slow
- Choice based on: seizure type/syndrome, age, sex (especially women of childbearing age), comorbidities, drug interactions, side effects
Drug Selection by Seizure Type
| Seizure Type | First-Line | Second-Line / Alternatives |
|---|---|---|
| Focal seizures | Lamotrigine, levetiracetam, carbamazepine, oxcarbazepine | Valproate, lacosamide, zonisamide, eslicarbazepine, perampanel |
| GTC (generalized) | Valproate, lamotrigine, levetiracetam | Topiramate, perampanel, zonisamide |
| Absence | Ethosuximide (drug of choice), valproate, lamotrigine | Clonazepam |
| Juvenile Myoclonic Epilepsy | Valproate (most effective), levetiracetam, lamotrigine | Topiramate, clonazepam; avoid carbamazepine/oxcarbazepine (worsen myoclonus) |
| Myoclonic seizures | Valproate, levetiracetam, clonazepam | Zonisamide, piracetam |
| Atonic / tonic (LGS) | Valproate, lamotrigine, rufinamide | Fenfluramine, clobazam, topiramate, ACTH |
| Infantile spasms (West) | ACTH / vigabatrin (tuberous sclerosis) | Prednisolone, pyridoxine |
| Dravet syndrome | Clobazam, valproate, stiripentol | Fenfluramine, cannabidiol (CBD); avoid carbamazepine/phenytoin |
Key Drug Details
| Drug | Mechanism | Key Features / Side Effects |
|---|---|---|
| Valproate | Na+ channel, GABA ↑, Ca2+ channel | Broad spectrum; teratogenic (neural tube defects, cognitive); weight gain, tremor, hepatotoxicity; avoid in women of childbearing age |
| Lamotrigine | Na+ channel | Broad spectrum; skin rash (Stevens-Johnson if titrated too fast); titrate slowly; relatively safe in pregnancy |
| Levetiracetam | SV2A synaptic vesicle protein | Broad spectrum; minimal drug interactions; behavioral side effects (irritability, depression) |
| Carbamazepine | Na+ channel | Focal seizures; enzyme inducer; hyponatremia; rash; teratogenic; worsen absence/myoclonus |
| Phenytoin | Na+ channel | Acute: IV for SE; chronic: gingival hyperplasia, ataxia, hirsutism, folate deficiency; zero-order kinetics |
| Ethosuximide | T-type Ca2+ channel (thalamus) | Absence only; GI side effects |
| Topiramate | Na+ channel, GABA, AMPA | Weight loss; cognitive impairment ("topiramate fog"); nephrolithiasis; teratogenic |
| Lacosamide | Slow inactivation of Na+ channel | Focal seizures; IV available; cardiac conduction |
| Zonisamide | Na+ + T-type Ca2+ channels | Broad spectrum; weight loss; nephrolithiasis |
| Perampanel | AMPA receptor antagonist | Focal and GTC; behavioral side effects |
| Cenobamate | Na+ channel + GABA-A | Drug-resistant focal epilepsy; Cochrane review 2024 confirms add-on efficacy |
| Fenfluramine | Serotonin/sigma-1 receptor | Dravet syndrome and LGS; FDA-approved |
| Cannabidiol (CBD, Epidiolex) | Multiple; unclear | Dravet, LGS, tuberous sclerosis complex |
Women and Epilepsy
- Valproate is contraindicated in women of childbearing potential where alternatives exist (teratogenic, cognitive effects on offspring)
- Lamotrigine is preferred in pregnancy (relative safety, but doses need adjustment as levels fall in pregnancy)
- All women with epilepsy taking antiseizure medications should take high-dose folic acid (5 mg/day)
Status Epilepticus (SE)
Definition
Seizure lasting ≥5 minutes OR two or more seizures without return to baseline consciousness.
Convulsive SE is a medical emergency with 10-20% mortality and risk of permanent neurological damage.
Management (Time-based Protocol)
| Time | Phase | Treatment |
|---|---|---|
| 0-5 min | Stabilisation | ABC, IV access, O2, glucose check, cardiac monitor |
| 5-20 min | Initial (benzodiazepine) | Lorazepam 0.1 mg/kg IV (or diazepam IV/rectal, or midazolam IM/buccal if no IV access) |
| 20-40 min | Second-line (if seizure continues) | Levetiracetam 60 mg/kg IV, or fosphenytoin 20 mg PE/kg IV, or valproate 40 mg/kg IV |
| 40-60 min | Third-line (refractory SE) | Anaesthetic doses: propofol, midazolam infusion, thiopental (ICU, intubation, continuous EEG monitoring) |
Always investigate for and treat the underlying cause (hypoglycemia, CNS infection, metabolic derangement, drug toxicity, structural lesion).
Drug-Resistant Epilepsy
Defined as failure of adequate trials of two tolerated, appropriately chosen antiseizure drugs. Affects ~30% of patients.
Surgical Options
- Temporal lobectomy / selective amygdalohippocampectomy: best results for MTLE with hippocampal sclerosis; 60-80% seizure-free
- Lesionectomy: resection of dysplasia, tumor, AVM
- Corpus callosotomy: palliative for atonic/tonic/GTC in LGS
- Hemispherotomy/hemispherectomy: for hemispheric epilepsy syndromes in children
Pre-surgical evaluation: Video-EEG, MRI, neuropsychology, FDG-PET, ictal SPECT (SISCOM), +/- intracranial EEG (stereo-EEG or subdural grids)
Neuromodulation
- Vagus nerve stimulation (VNS): reduces seizure frequency by ~50% in ~50% of patients; not curative
- Responsive neurostimulation (RNS): closed-loop cortical stimulation detecting and aborting seizure onset
- Deep brain stimulation (DBS of anterior nucleus of thalamus): effective for refractory focal epilepsy
Dietary Therapy
- Ketogenic diet (KD): high fat, very low carbohydrate; effective for drug-resistant epilepsy especially in children, Dravet syndrome, glucose transporter deficiency (GLUT1); mechanism: ketone bodies reduce neuronal excitability
Differential Diagnosis of Seizures
| Condition | Key distinguishing features |
|---|---|
| Syncope | Prodrome (light-headedness, nausea, visual grey-out); brief (<30 sec); triggered (standing, pain); no prolonged postictal |
| TIA | Negative symptoms (weakness, sensory loss); not convulsive; brief |
| Psychogenic non-epileptic seizures (PNES) | Emotional trigger; eyes closed; pelvic thrusting; long duration; no postictal; normal ictal EEG (video-EEG confirms) |
| Migraine with aura | Slow march of visual/sensory aura over minutes; followed by headache |
| Hypoglycemia | Check blood glucose; tremor, sweating, confusion |
| Transient global amnesia | Sudden amnesia; repetitive questioning; no motor features |
| Breath-holding spell | Children; triggered by crying/frustration; cyanotic vs. pallid type |
| Sleep disorders | REM behavior disorder, night terrors, parasomnias |
Cognitive and Psychiatric Comorbidities
Epilepsy is associated with significant cognitive and psychiatric comorbidity - not merely epiphenomena but a bidirectional relationship:
-
Memory deficits are the most common, particularly in temporal lobe epilepsy; left TLE causes verbal memory impairment
-
Executive dysfunction and visuoconstructional deficits occur in both left and right TLE
-
Depression (affects ~30-40%); anxiety; psychosis (particularly postictal or interictal)
-
SUDEP (Sudden Unexpected Death in Epilepsy) - major cause of premature mortality; risk increased with uncontrolled seizures, nocturnal GTC, young men
-
Social impact: driving restrictions, employment limitations, stigma
-
Harrison's 22E, Ch. 436 | Bradley & Daroff's Neurology
Meningitis
Finding Sources
Searching PubMed
"meningitis bacterial"[MeSH Terms] AND "treatment"[MeSH Terms]
Finding Sources
Searching PubMed
"meningitis"[MeSH Terms] AND "dexamethasone"[MeSH Terms]
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive, detailed information from Harrison's 22E covering bacterial and viral meningitis in full. Let me compile the complete answer.
Meningitis
Definition
Meningitis is inflammation of the arachnoid membrane, pia mater, and the intervening cerebrospinal fluid (CSF), extending throughout the subarachnoid space around the brain, spinal cord, and into the ventricles.
- Bacterial (pyogenic) meningitis: acute, polymorphonuclear (PMN) CSF response; a medical emergency
- Viral (aseptic) meningitis: usually self-limiting; lymphocytic CSF; most common form
- Tuberculous/fungal/carcinomatous: subacute to chronic course
Epidemiology
-
Bacterial meningitis incidence: ~1.4 cases/100,000/year in the US
-
Community-acquired causes:
- Streptococcus pneumoniae ~50%
- Neisseria meningitidis ~25%
- Group B Streptococcus ~15%
- Listeria monocytogenes ~10%
- Haemophilus influenzae type b: <10% (markedly reduced post-vaccination)
-
Mortality: ~15-20% despite antibiotic therapy
-
N. meningitidis causes recurring epidemics every 8-12 years
-
Harrison's 22E | Goldman-Cecil Medicine
Etiology by Age/Risk Group
| Patient Group | Common Organisms |
|---|---|
| Neonates (<1 month) | Group B Streptococcus, E. coli, Listeria monocytogenes, Klebsiella |
| Infants 1-3 months | Above + S. pneumoniae, N. meningitidis |
| Children >3 months - adults <55 | S. pneumoniae, N. meningitidis |
| Adults >55, alcoholism, chronic illness | S. pneumoniae, Listeria, gram-negative bacilli |
| Immunocompromised | Listeria, gram-negative bacilli, Cryptococcus neoformans, CMV |
| Post-neurosurgery/VP shunt | Staphylococci (S. aureus, coagulase-negative), gram-negatives (Pseudomonas) |
| Otitis/sinusitis/mastoiditis | Streptococcus spp., Haemophilus, gram-negative anaerobes |
| Endocarditis | Viridans streptococci, S. aureus, HACEK organisms |
| Complement deficiency/properdin | N. meningitidis (recurrent) |
Listeria is acquired through contaminated food (coleslaw, soft cheeses, deli meats, hot dogs). - Harrison's 22E
Pathophysiology
- Nasopharyngeal colonization → S. pneumoniae and N. meningitidis attach to and invade nasopharyngeal epithelium
- Bacteremia → polysaccharide capsule resists phagocytosis and complement-mediated killing
- CNS invasion → bacteria enter via choroid plexus or cerebral capillary endothelium → replicate freely in CSF (normal CSF has very few WBCs, little complement or immunoglobulin)
- Inflammatory cascade → bacterial components (LPS from gram-negatives, teichoic acid/peptidoglycan from gram-positives) trigger cytokines (TNF-α, IL-1β, IL-6) → blood-brain barrier disruption → cerebral edema, vasculitis, thrombosis, raised ICP
Clinical Features
Classic Triad (present in only ~44% of patients on presentation)
- Fever
- Neck stiffness (nuchal rigidity)
- Altered consciousness / headache
Full Spectrum of Symptoms
- Severe headache (most constant symptom)
- Fever, rigors
- Photophobia and phonophobia
- Neck stiffness - resist passive neck flexion
- Nausea, vomiting
- Seizures (~20-40%)
- Altered consciousness - confusion → stupor → coma
- Focal neurological deficits (cranial nerve palsies, hemiparesis) in ~20%
Meningeal Signs
- Kernig's sign: With hip flexed at 90°, inability to extend the knee beyond 135° without pain
- Brudzinski's sign: Passive neck flexion causes involuntary flexion of the hips and knees
- Jolt accentuation: Worsening of headache with rapid horizontal head rotation (sensitive but not specific)
Specific Features by Organism
| Organism | Distinguishing Features |
|---|---|
| N. meningitidis | Petechial/purpuric rash (non-blanching); fulminant course; Waterhouse-Friderichsen syndrome (bilateral adrenal hemorrhage); DIC |
| S. pneumoniae | Often with pneumonia, sinusitis, otitis; alcoholism/splenectomy; highest mortality |
| Listeria | Brainstem involvement (rhombencephalitis); elderly/immunocompromised; pregnancy |
| Herpes simplex | Encephalitic features (temporal lobe involvement); behavioral change; seizures |
| Tuberculosis | Subacute onset; cranial nerve palsies; basal exudates; hydrocephalus |
| Cryptococcus | Immunocompromised (HIV); subacute/chronic; positive India ink; elevated opening pressure |
CSF Analysis - The Key Investigation
Lumbar puncture (LP) is the definitive diagnostic test. Perform CT head first if: focal neurological deficit, papilloedema, new seizure, reduced consciousness, or immunocompromised.
CSF Findings Comparison
| Parameter | Normal | Bacterial | Viral | TB/Fungal |
|---|---|---|---|---|
| Appearance | Clear | Turbid/cloudy | Clear | Clear/xanthochromic |
| Opening pressure | <180 mmH₂O | ↑↑ (>180 in 90%) | Normal/mildly ↑ | ↑ |
| WBC | <5/μL | PMN >100/μL (90%) | Lymphocytes 25-500/μL | Lymphocytes (early PMN) |
| Glucose | 2.8-4.4 mmol/L | ↓↓ <2.2 mmol/L (60%) | Normal/slightly ↓ | ↓↓ |
| CSF:serum glucose | >0.6 | <0.4 (highly suggestive) | >0.6 | <0.4 |
| Protein | <0.45 g/L | ↑ >0.45 g/L (90%) | Normal/slightly ↑ | ↑↑ |
| Gram stain | Negative | Positive >60% | Negative | Negative (AFB positive in ~10-40%) |
| Culture | Negative | Positive >70% | Negative | Positive in weeks |
Additional CSF Tests
- CSF multiplex PCR (BioFire FilmArray panel): detects S. pneumoniae, N. meningitidis, L. monocytogenes, HSV-1/2, CMV, EBV, enterovirus, cryptococcus - rapid result
- Cryptococcal antigen: for immunocompromised patients
- India ink preparation: Cryptococcus (positive in ~50%)
- AFB smear and culture: TB meningitis
- Adenosine deaminase (ADA): elevated in TB meningitis
- Cytology and flow cytometry: carcinomatous/lymphomatous meningitis
- Lactate: >3.5 mmol/L favors bacterial meningitis
Serum Biomarkers
- Procalcitonin: elevated in bacterial; helps distinguish from viral
- Blood cultures: positive in ~50-80% of bacterial meningitis
- CBC, CRP, ESR
Investigations Summary
| Test | Purpose |
|---|---|
| CT head (before LP if indicated) | Exclude mass lesion, cerebral herniation risk |
| LP + CSF analysis | Definitive diagnosis (opening pressure, cell count, differential, glucose, protein, culture, PCR, Gram stain) |
| Blood cultures (x2) | Before antibiotics; positive in majority |
| Bloods: FBC, CMP, coagulation, LFTs, glucose | Assess severity, metabolic status |
| Procalcitonin / CRP | Bacterial vs. viral discrimination |
| Meningococcal PCR (blood/CSF) | N. meningitidis in rash with purpura |
| Chest X-ray / CT chest | Source (pneumonia, TB) |
| MRI brain | Complications: cerebral edema, infarction, abscess, empyema, hydrocephalus |
Treatment
Step 1: Immediate Empirical Antibiotics
Bacterial meningitis is a medical emergency. Target antibiotic administration within 60 minutes of arrival.
Do NOT delay antibiotics for CT or LP if there is clinical suspicion. Draw blood cultures first, then give antibiotics.
Empirical Antibiotic Regimens by Age/Risk
| Patient Group | Empirical Regimen |
|---|---|
| Preterm/neonates <1 month | Ampicillin + cefotaxime |
| Infants 1-3 months | Ampicillin + cefotaxime or ceftriaxone |
| Immunocompetent children >3 months and adults <55 | Cefotaxime or ceftriaxone + vancomycin |
| Adults >55 / alcoholism / debilitating illness | Ampicillin + cefotaxime or ceftriaxone + vancomycin (adds Listeria coverage) |
| Hospital-acquired / post-neurosurgery / neutropenic / immunocompromised | Ampicillin + ceftazidime or meropenem + vancomycin |
Add:
- Acyclovir (empirically) - HSV encephalitis is the leading differential
- Doxycycline during tick season (rickettsial / Ehrlichia)
- Metronidazole if source is otitis/sinusitis/mastoiditis (gram-negative anaerobes)
Why vancomycin? Due to emergence of penicillin- and cephalosporin-resistant S. pneumoniae
Drug Doses (Adults)
| Drug | Adult Dose |
|---|---|
| Ampicillin | 12 g/day IV, q4h |
| Cefotaxime | 12 g/day IV, q4h |
| Ceftriaxone | 4 g/day IV, q12h |
| Vancomycin | 45-60 mg/kg/day IV, q8-12h (target AUC 400-600) |
| Acyclovir | 10 mg/kg IV, q8h |
Step 2: Dexamethasone (Adjunctive Therapy)
- Dexamethasone 0.15 mg/kg q6h for 4 days, given 10-20 minutes BEFORE or with the first dose of antibiotics
- Mechanism: reduces cytokine-mediated inflammation; decreases BBB disruption
- Benefit: reduces hearing loss and neurological complications, especially in S. pneumoniae meningitis
- Evidence: landmark trial (de Gans and van de Beek, NEJM 2002) showed reduced mortality and morbidity for pneumococcal meningitis in adults
- Caveat: may reduce vancomycin penetration into CSF - ensure adequate doses; if penicillin-sensitive pneumococcus confirmed, stop vancomycin
Step 3: Targeted Therapy (Once Organism Identified)
| Organism | Drug of Choice | Alternative |
|---|---|---|
| S. pneumoniae (penicillin-sensitive, MIC <0.1) | Penicillin G 24 million U/day | Ceftriaxone |
| S. pneumoniae (resistant) | Ceftriaxone + vancomycin | Meropenem |
| N. meningitidis | Penicillin G or ceftriaxone | Chloramphenicol |
| L. monocytogenes | Ampicillin (+ gentamicin in severe cases) | TMP-SMX |
| H. influenzae | Ceftriaxone | Chloramphenicol |
| S. aureus (MSSA) | Nafcillin/flucloxacillin | Vancomycin |
| S. aureus (MRSA) | Vancomycin ± rifampicin | Linezolid |
| Gram-negative bacilli / Pseudomonas | Ceftazidime or meropenem | Cefepime |
| Cryptococcus neoformans | Liposomal amphotericin B + flucytosine (induction x2 weeks), then fluconazole | |
| TB meningitis | RIPE (Rifampicin + Isoniazid + Pyrazinamide + Ethambutol) + dexamethasone |
Duration of Antibiotics
- N. meningitidis: 7 days
- H. influenzae: 7-10 days
- S. pneumoniae: 10-14 days
- L. monocytogenes: ≥21 days
- Gram-negative bacilli: 21 days
Viral Meningitis (Aseptic Meningitis)
Etiology (most common to less common):
- Enteroviruses (coxsackieviruses, echoviruses) - most common; seasonal (summer/fall)
- HSV-2 > HSV-1
- Varicella-zoster virus (VZV)
- EBV, CMV, HHV-6
- Arboviruses (West Nile virus, others) - note: WNV can cause PMN pleocytosis (45%)
- HIV (seroconversion illness)
- Mumps (in unvaccinated populations)
CSF: Lymphocytic pleocytosis (25-500/μL); normal glucose; normal to mildly elevated protein; negative Gram stain; identify by PCR
Treatment:
- Mostly supportive (rest, analgesia, antipyretics, hydration)
- Acyclovir for HSV or VZV meningitis (IV acyclovir 10 mg/kg q8h for moderate-severe; oral for mild VZV)
- Antiretrovirals for HIV seroconversion meningitis
- Enteroviruses: Pleconaril (investigational; not widely available); generally self-limiting
Prognosis: Excellent; full recovery in the vast majority within 1-2 weeks.
Complications of Bacterial Meningitis
| Complication | Mechanism | Features |
|---|---|---|
| Raised ICP | Cerebral edema, hydrocephalus | Headache, vomiting, papilloedema, Cushing's triad, herniation |
| Cerebral infarction | Vasculitis, thrombosis, vasospasm | Focal deficit, hemiplegia |
| Seizures | Cortical irritation, infarction | 20-40% of cases |
| Hydrocephalus | CSF obstruction by exudates | Worsening consciousness |
| Subdural empyema / brain abscess | Extension of infection | Focal signs, worsening despite antibiotics |
| Cranial nerve palsies | Basal exudates | CN III, IV, VI, VII, VIII most common |
| Sensorineural hearing loss | Most common neurological sequel; pneumococcal > others | Up to 30% with S. pneumoniae - reduced by dexamethasone |
| DIC / Waterhouse-Friderichsen | N. meningitidis; endotoxin-mediated | Purpura, adrenal hemorrhage, shock |
| SIADH | Inappropriate ADH secretion | Hyponatraemia; fluid restrict |
| Septic shock | Bacteraemia | Haemodynamic instability |
Chemoprophylaxis for Close Contacts
Indicated for close contacts of N. meningitidis cases:
- Rifampicin 600 mg q12h for 2 days (adults); or
- Ciprofloxacin 500 mg single dose (adults); or
- Ceftriaxone 250 mg IM single dose
Not indicated for S. pneumoniae meningitis contacts.
Vaccination: Meningococcal conjugate vaccine (serogroups A, C, W-135, Y) + serogroup B vaccine for close contacts and high-risk groups (complement deficiency, asplenia, complement inhibitor therapy).
Differential Diagnosis of Meningitis
| Condition | Key distinguishing features |
|---|---|
| Subarachnoid haemorrhage | "Thunderclap" headache; xanthochromic CSF; CT shows blood |
| Brain abscess | Focal deficit; less meningism; ring-enhancing lesion on MRI |
| Encephalitis | Prominent psychiatric/cognitive features; seizures; focal signs > meningism |
| Neurosarcoidosis | Chronic; cranial nerve palsies; hilar lymphadenopathy; CSF ACE |
| Carcinomatous meningitis | Malignancy history; multiple cranial nerve palsies; positive CSF cytology |
| Drug-induced aseptic meningitis | NSAIDs, TMP-SMX, IVIG; takes weeks-months |
| Migraine | No fever; normal CSF; recurrent; responds to triptans |
Summary: Approach to Suspected Meningitis
Suspected meningitis
├── Stabilise: ABC, IV access, O2, blood cultures
├── Any contraindication to LP? (focal deficit, papilloedema, new seizure, coma, immunocompromised)
│ ├── YES → CT head first → LP when safe
│ └── NO → LP immediately
├── Give antibiotics within 60 min regardless of LP result
├── Dexamethasone BEFORE or WITH first antibiotic dose
├── Add acyclovir (exclude HSV encephalitis)
└── Adjust therapy when CSF/culture results available
Neuroimaging
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
This is a shared conversation. Sign in to Orris to start your own chat.