Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism diagram
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive material from multiple authoritative textbooks. Let me compile the complete explanation.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition & Berlin Criteria
ARDS is a clinical syndrome of severe, rapidly progressive hypoxemic respiratory failure caused by diffuse alveolar damage (DAD). The 2012 Berlin Definition classifies it by PaO₂/FiO₂ ratio with ≥5 cmH₂O PEEP:
- Mild: 200–300 mmHg
- Moderate: 100–200 mmHg
- Severe: <100 mmHg
Additional criteria: bilateral infiltrates on CXR/CT not fully explained by cardiac failure or fluid overload, onset within 7 days of a known clinical insult. — Harrison's Principles of Internal Medicine, 22E
Causes: Direct vs. Indirect Lung Injury
| Direct Lung Injury | Indirect Lung Injury |
|---|---|
| Pneumonia | Sepsis (most common: 40–60%) |
| Aspiration of gastric contents | Severe trauma / multiple fractures |
| Pulmonary contusion | Burns, pancreatitis |
| Near-drowning, toxic inhalation | Multiple transfusions, drug overdose |
Pathogenesis: Step-by-Step Mechanism
1. Initiating Insult → Innate Immune Activation
Whether the injury is direct (inhaled pathogens, aspiration) or indirect (systemic sepsis, trauma), the trigger activates pattern recognition receptors (e.g., Toll-like receptors) on alveolar type I (ATI) epithelial cells and resident alveolar macrophages. This initiates a downstream inflammatory cascade. — Harrison's, Fig. 312-3
2. Cytokine Storm & Neutrophil Recruitment
Activated macrophages release a cascade of pro-inflammatory mediators:
- TNF-α, IL-1β, IL-6, IL-8 → systemic and local inflammation
- IL-8 and other PMN chemokines → massive recruitment of polymorphonuclear neutrophils (PMNs) from the pulmonary capillaries
Neutrophils are the central effectors of injury. They marginate in alveolar capillaries and transmigrate across the endothelium and epithelium into the alveolar space. Once there, they release:
- Proteases (elastase, matrix metalloproteinases)
- Reactive oxygen species (ROS)
- Neutrophil extracellular traps (NETs)
- Platelet-activating factor (PAF)
These products directly destroy both the endothelial barrier and the alveolar epithelium. — Robbins & Kumar Basic Pathology; Harrison's; Murray & Nadel's
3. Breakdown of the Alveolar-Capillary Barrier
Normally, the alveolar-capillary membrane is virtually impermeable to proteins. In ARDS:
- Tight junctions between endothelial cells are disrupted ("disrupted tight junctions")
- ATI pneumocytes (which cover ~95% of the alveolar surface) are necrosed
- Na⁺/K⁺-ATPase and ENaC (epithelial sodium channels) on ATII cells are inactivated → impaired fluid and ion clearance
The result is high-permeability (non-cardiogenic) pulmonary edema: protein-rich, inflammatory fluid floods the alveolar space. This is the defining pathophysiological event. — Harrison's, Fig. 312-3; Fishman's Pulmonary Diseases
4. Surfactant Dysfunction & Alveolar Collapse
- ATII pneumocytes are damaged and their lamellar bodies (surfactant storage organelles) are disrupted
- Phospholipase A₂ (released during pancreatic injury, or by inflammatory cells) enzymatically degrades surfactant
- Protein-rich edema fluid further inactivates surfactant
Loss of surfactant dramatically increases alveolar surface tension → diffuse microatelectasis and reduced lung compliance.
5. Hyaline Membrane Formation
Proteins that leak into the alveoli (fibrin, plasma proteins) precipitate along the denuded alveolar walls to form hyaline membranes — the pathognomonic histological finding. These membranes:
- Further impair gas exchange
- Serve as a scaffold for subsequent fibroproliferation
6. Pulmonary Microvascular Thrombosis
- PMN-platelet aggregates and monocyte-platelet aggregates form in the microvasculature
- Fibrin deposition causes microvascular obstruction
- Endothelial injury promotes a pro-coagulant state
- This increases pulmonary dead space (ventilated but not perfused alveoli) and can lead to pulmonary hypertension
The Three Phases of ARDS

Phase 1: Exudative (Days 0–7)
- Diffuse alveolar damage, alveolar flooding with protein-rich edema
- Neutrophil-predominant infiltrate
- Hyaline membrane formation
- Severe hypoxemia (↓ V/Q matching, true intrapulmonary shunt)
- Reduced FRC, reduced lung compliance
Phase 2: Proliferative (Days 7–21)
- ATII pneumocytes proliferate along denuded basement membranes → replace ATI cells and differentiate into new ATI pneumocytes
- Shift from neutrophil- to lymphocyte-predominant infiltrate
- Organization of alveolar exudates; early fibrotic changes
- Most patients begin to improve, though dyspnea/hypoxemia persist
Phase 3: Fibrotic (Day 21+, ~20% of patients)
- Alveolar exudates converted to dense alveolar-duct and interstitial fibrosis
- Vascular intimal fibroproliferation → pulmonary hypertension
- Emphysema-like bullae; increased risk of pneumothorax
- Severely impaired compliance and diffusion capacity; high dead space
The Injured Alveolus — Summary Diagram
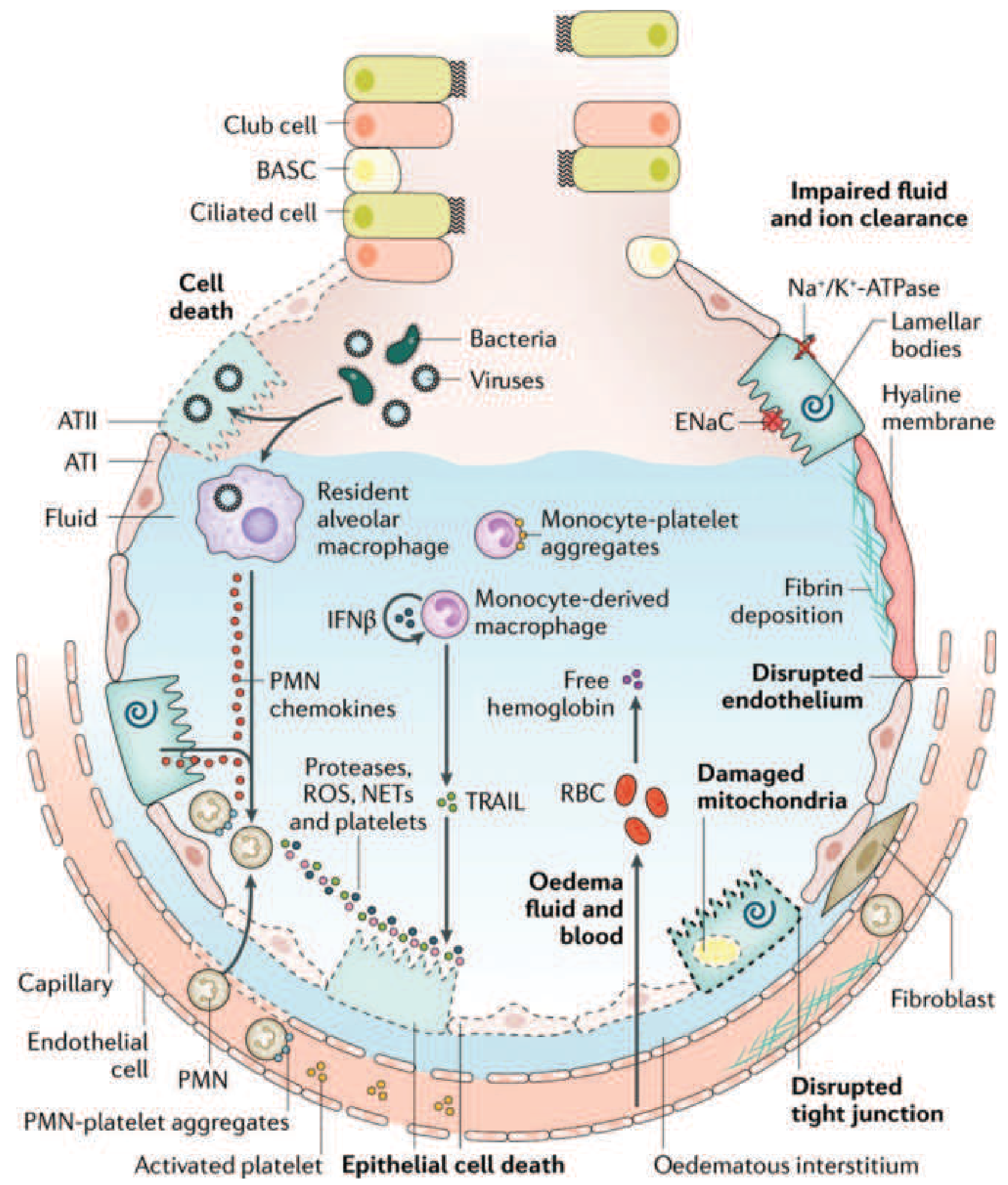
Harrison's Fig. 312-3 — The injured alveolus in the acute phase of ARDS
Physiological Consequences
| Mechanism | Consequence |
|---|---|
| Alveolar flooding + microatelectasis | ↓ FRC, intrapulmonary shunt, refractory hypoxemia |
| Surfactant loss | ↓ Compliance ("stiff lungs") |
| Microvascular obstruction | ↑ Dead space, pulmonary hypertension |
| Impaired Na⁺/K⁺-ATPase on ATII cells | Impaired edema clearance |
| Hyaline membranes | Diffusion barrier, scaffold for fibrosis |
The resulting hypoxemia is characteristically refractory to supplemental O₂ alone because it is driven by true shunt (blood traversing completely unventilated alveoli) rather than V/Q mismatch.
Ventilator-Induced Lung Injury (VILI) — Secondary Mechanism
Mechanical ventilation, while necessary, can worsen injury through:
- Volutrauma/barotrauma: Overdistension of relatively "normal" lung units adjacent to consolidated areas
- Atelectrauma: Cyclic collapse and re-expansion of unstable alveoli
- Both mechanisms amplify cytokine release and worsen barrier disruption
This is the rationale for lung-protective ventilation (tidal volume 6 mL/kg IBW, plateau pressure ≤30 cmH₂O). — Harrison's; Fishman's, Fig. 140-3
Sources: Harrison's Principles of Internal Medicine 22E; Murray & Nadel's Textbook of Respiratory Medicine; Fishman's Pulmonary Diseases and Disorders; Robbins & Kumar Basic Pathology; Sabiston Textbook of Surgery
This is a shared conversation. Sign in to Orris to start your own chat.