Thalassemia make every important points
Reading File
Finding Sources
Finding Sources
Searching Images
thalassemia blood smear clinical features
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Here is a comprehensive, exam-ready breakdown of all the important points on Thalassemia.
Thalassemia — Complete Overview
1. Definition
Thalassemia is a genetically heterogeneous disorder caused by germline mutations that decrease synthesis of either α-globin or β-globin chains, leading to:
- Hemoglobin deficiency → hypochromic, microcytic anemia
- Imbalance in globin chain synthesis → excess unpaired chains → intracellular precipitates → hemolysis
- Two mechanisms of anemia: decreased red cell production (ineffective erythropoiesis) + decreased red cell lifespan (hemolysis)
"Thalassa" means "sea" in Greek — the disease is named for its prevalence around the Mediterranean.
— Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 602
2. Genetics & Chromosomal Location
| Globin Chain | Gene Location | Type of Mutation |
|---|---|---|
| β-globin | Chromosome 11 (single gene) | Mainly point mutations |
| α-globin | Chromosome 16 (two identical genes in tandem = 4 total) | Mainly gene deletions |
Inheritance: Autosomal codominant
3. Epidemiology
- Endemic in the Mediterranean basin, Middle East, tropical Africa, Indian subcontinent, Asia
- Among the most common inherited disorders of humans
- High prevalence explained by protection against falciparum malaria in heterozygous carriers (similar to HbS)
4. β-Thalassemia
Molecular Pathogenesis
Over 100 causative mutations identified, divided into:
| Mutation Type | Effect | Result |
|---|---|---|
| β⁰ | No β-globin synthesized | Absent chain production |
| β⁺ | Reduced β-globin synthesized | Decreased (not absent) chain production |
Three major classes:
- Splicing mutations — Most common cause of β⁺-thalassemia. Destroy normal RNA splice junctions (→ β⁰) or create ectopic splice sites (→ β⁺, since some normal mRNA still made)
- Promoter region mutations — Reduce transcription by 75–80%; always → β⁺
- Chain terminator mutations — Most common cause of β⁰-thalassemia; nonsense mutations (premature stop codon) or frameshift mutations (small insertions/deletions) block translation entirely
Pathogenesis of Anemia (Two Mechanisms)
- Reduced HbA synthesis → hypochromic, microcytic RBCs with low oxygen-carrying capacity
- Excess unpaired α-chains precipitate within RBC precursors → insoluble inclusions → membrane damage → apoptosis of RBC precursors (ineffective erythropoiesis) + hemolysis of mature RBCs in spleen
Clinical Classification
| Syndrome | Genotype | Clinical Features |
|---|---|---|
| β-Thalassemia Major | Homozygous (β⁰/β⁰, β⁺/β⁰, β⁺/β⁺) | Severe anemia; requires regular blood transfusions |
| β-Thalassemia Intermedia | Variable | Moderately severe; transfusions not required |
| β-Thalassemia Minor (Trait) | Heterozygous (β⁰/β, β⁺/β) | Asymptomatic/mild anemia; red cell abnormalities on smear |
5. β-Thalassemia Major (Cooley's Anemia) — Key Features
Onset: Symptoms appear 6–9 months after birth (when HbF → HbA switch occurs)
Clinical Features:
- Severe microcytic hypochromic anemia with hemoglobin as low as 3–4 g/dL
- Hepatosplenomegaly — from extramedullary hematopoiesis + RBC destruction
- Expansion of the erythroid marrow → skeletal changes:
- "Chipmunk facies" — maxillary hyperplasia, prominent cheekbones
- Frontal bossing
- "Crew-cut" appearance on skull X-ray — from hair-on-end striations
- Thinning of cortical bone → pathological fractures
- Growth retardation
- Iron overload (secondary hemochromatosis) — from chronic transfusions + increased GI absorption
- Organ damage: cardiac failure (most common cause of death), liver cirrhosis, endocrine failure (diabetes, hypogonadism)
- Jaundice (unconjugated hyperbilirubinemia)
- Gallstones (pigment stones from chronic hemolysis)
"Thalassemic Facies"
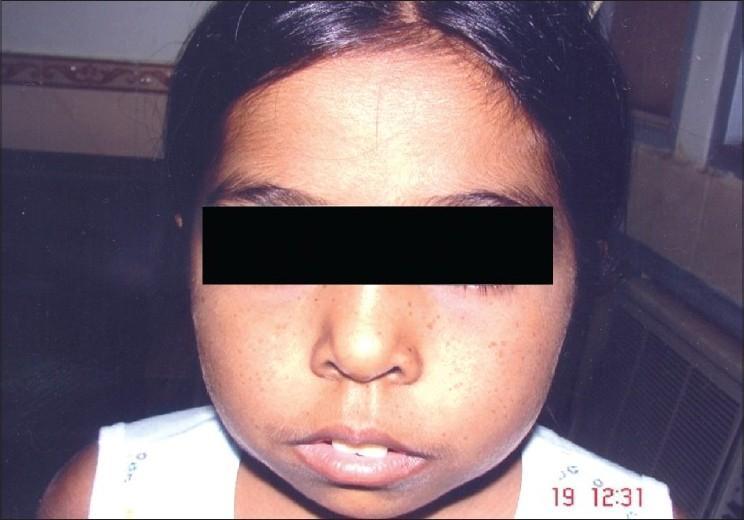
Classic facial features of β-thalassemia major: frontal bossing, maxillary hyperplasia, flattened nasal bridge ("chipmunk facies") from bone marrow expansion.
Hepatosplenomegaly + Peripheral Smear
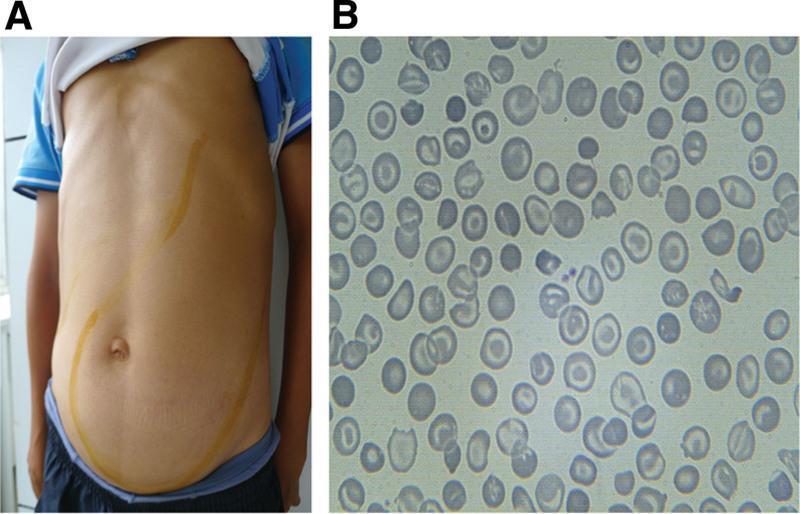
A: Massive hepatosplenomegaly from extramedullary hematopoiesis. B: Peripheral smear showing marked microcytosis, hypochromia, target cells (codocytes), and schistocytes.
Lab Findings:
- ↓ Hb, ↓ MCV, ↓ MCH
- Target cells, microcytes, hypochromic cells, nucleated RBCs, basophilic stippling on smear
- ↑ Reticulocytes (modest — less than expected due to ineffective erythropoiesis)
- ↑ Serum iron, ↑ ferritin
- ↑ HbF (compensatory); ↑ HbA2 (in β-thalassemia minor)
- Absent or ↓ HbA
Peripheral Blood Smear (Hb E/β-thalassemia)
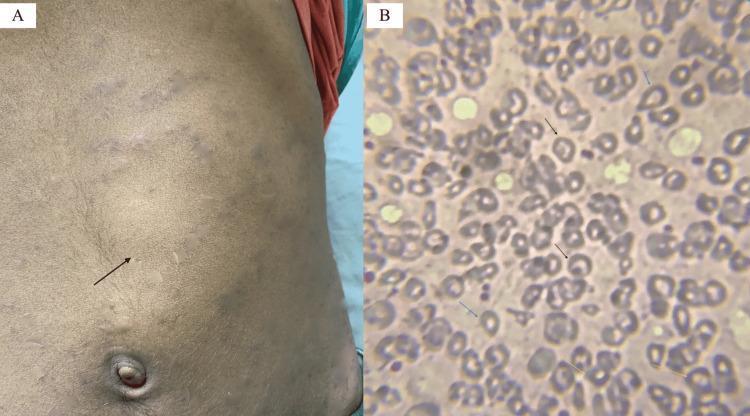
Leishman stain (200×): target cells, teardrop cells, and basophilic stippling — classic findings in thalassemia.
6. β-Thalassemia Minor (Trait)
- Heterozygous: one normal + one mutated β-globin allele
- Asymptomatic — incidental finding
- Mild microcytic hypochromic anemia (Hb 9–11 g/dL)
- ↑ HbA2 (>3.5%) — diagnostic hallmark
- Mild ↑ HbF possible
- No treatment required; important for genetic counseling
7. α-Thalassemia
Caused by deletion of α-globin genes (usually). Severity depends on how many of the 4 α-globin genes are deleted.
| Syndrome | Genotype | Features |
|---|---|---|
| Silent carrier | −/α, α/α (1 gene deleted) | Asymptomatic; slight microcytosis only |
| α-Thalassemia trait | −/−, α/α or −/α, −/α (2 genes deleted) | Asymptomatic; resembles β-thal minor; normal HbA2 |
| HbH disease | −/−, −/α (3 genes deleted) | Moderately severe anemia; resembles β-thal intermedia |
| Hydrops fetalis (α-thal major) | −/−, −/− (4 genes deleted) | Lethal in utero or at birth |
HbH Disease
- Only 1 α-globin gene functioning → excess β-chains form tetramers (HbH = β₄)
- HbH has extremely high O₂ affinity → delivers little O₂ to tissues
- HbH oxidizes easily → precipitates → RBC inclusions → sequestration in spleen
- Moderately severe hemolytic anemia
Hydrops Fetalis (α-Thalassemia Major)
- All 4 α-globin genes deleted → excess γ-chains form Hb Bart's (γ₄)
- Hb Bart's has very high O₂ affinity → severe fetal tissue hypoxia
- Presents in third trimester with:
- Severe pallor
- Generalized edema (hydrops)
- Massive hepatosplenomegaly
- Historically fatal; intrauterine transfusion now saves many infants
- Lifelong transfusion dependence → risk of iron overload
- Hematopoietic stem cell transplantation (HSCT) can be curative
Ethnic note:
- Asian populations: −/− haplotype common → at risk for HbH disease and hydrops fetalis
- African populations: −/α, −/α genotype → symptomatic α-thal is rare
8. Compensatory Mechanisms & Complications
| Mechanism | Consequence |
|---|---|
| Extramedullary hematopoiesis (liver, spleen, lymph nodes, bones) | Hepatosplenomegaly, skeletal deformities |
| Expansion of erythroid marrow | Facial/skeletal changes, osteoporosis |
| Increased GI iron absorption | Iron overload even without transfusions |
| Transfusion therapy | Secondary hemochromatosis → cardiac failure, cirrhosis, endocrinopathy |
| ↑ HbF production | Partially compensates in β-thalassemia; HbF does not require β-chains |
9. Diagnosis
| Test | Findings |
|---|---|
| CBC | ↓ Hb, ↓ MCV (microcytic), ↓ MCH (hypochromic) |
| Peripheral smear | Target cells, microcytes, nucleated RBCs, basophilic stippling, teardrop cells |
| Hemoglobin electrophoresis / HPLC | ↑ HbA2 (β-thal minor), ↑ HbF (major), absent HbA (major), HbH or Hb Bart's (α-thal) |
| Serum iron/ferritin | Elevated (unlike iron deficiency) |
| DNA analysis / PCR | Confirms specific mutation; used in prenatal diagnosis |
| Bone marrow | Erythroid hyperplasia |
| Prenatal | Chorionic villus sampling (CVS) or amniocentesis + DNA analysis |
Key distinction from iron deficiency anemia:
- In thalassemia: ↑ serum iron, ↑ ferritin, ↑ HbA2
- In iron deficiency: ↓ serum iron, ↓ ferritin, normal HbA2
10. Treatment
| Treatment | Indication |
|---|---|
| Regular blood transfusions | β-Thalassemia major (every 3–4 weeks to keep Hb >9 g/dL) |
| Iron chelation therapy (deferoxamine, deferasirox, deferiprone) | Prevent iron overload from transfusions |
| Hematopoietic stem cell transplantation (HSCT) | Curative; best results in young patients without organ damage |
| Splenectomy | Reduces transfusion requirements; risk of post-splenectomy sepsis (vaccinate against encapsulated organisms) |
| Folic acid | Supports increased erythropoiesis |
| Hydroxyurea | Increases HbF production (especially in β-thalassemia intermedia) |
| Gene therapy / gene editing | Emerging; luspatercept (activin receptor ligand trap) approved to reduce transfusion burden |
| Intrauterine transfusion | Hydrops fetalis (α-thal major) |
11. Thalassemia & Malaria
- Heterozygous carriers are protected against Plasmodium falciparum malaria — explains high gene frequency in malaria-endemic regions (similar selective advantage as HbS in sickle cell trait)
12. Genetic Counseling
- Both parents β-thalassemia minor → 25% risk of major, 50% risk of minor, 25% normal
- Prenatal diagnosis by DNA analysis (CVS at 10–12 weeks or amniocentesis at 15–18 weeks)
- Important in populations of Mediterranean, Middle Eastern, South Asian, and Southeast Asian ancestry
Sources: Robbins, Cotran & Kumar — Pathologic Basis of Disease, pp. 602–611 | Robbins & Kumar — Basic Pathology, pp. 380–388
This is a shared conversation. Sign in to Orris to start your own chat.