pheochromocytoma
Reading File
Finding Sources
Searching PubMed
pheochromocytoma diagnosis management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive information to compose a detailed answer. Let me put it all together.
Pheochromocytoma
Definition and Overview
Pheochromocytomas are neoplasms of chromaffin cells of the adrenal medulla that synthesize and release catecholamines (epinephrine, norepinephrine, and sometimes dopamine). They are rare - estimated incidence of 2-8 cases per million per year - but clinically important because they cause a surgically correctable form of hypertension and can trigger fatal hypertensive crises if unrecognized.
When the same tumor arises at extraadrenal sites along sympathetic ganglia, it is called a paraganglioma.
The "Rule of 10s" (Historical)
| Feature | Proportion |
|---|---|
| Extraadrenal location | ~10% |
| Bilateral | ~10% (up to 50% in familial syndromes) |
| Malignant | ~10% (up to 20% in extraadrenal) |
| Not associated with hypertension | ~10% |
| Familial | >10% (higher than once thought) |
| Metastatic at presentation | >10% |
The rule has been modified to apply primarily to sporadic (non-familial) cases. In practice, familial/bilateral/metastatic rates are higher than the rule implies.
Pathogenesis and Genetics
Pheochromocytomas are genetically heterogeneous, with driver mutations in at least a dozen genes:
| Gene/Pathway | Syndrome | Mechanism |
|---|---|---|
| RET proto-oncogene | MEN2A, MEN2B | Activates growth factor receptor signaling |
| NF1 tumor suppressor | Neurofibromatosis type 1 | Growth factor receptor dysregulation |
| VHL tumor suppressor | Von Hippel-Lindau disease | Activates hypoxia-inducible factors (HIFs) |
| SDHB, SDHD | Familial paraganglioma | Succinate dehydrogenase complex mutations → HIF activation |
| EPAS1 | Sporadic | HIF pathway activation |
Associated syndromes in detail:
- MEN2A: Pheochromocytoma (~50%, usually bilateral) + medullary thyroid carcinoma + parathyroid adenomas. Associated with RET mutations.
- MEN2B: Bilateral pheochromocytoma + medullary thyroid carcinoma + submucosal neuromas + marfanoid habitus.
- Von Hippel-Lindau (VHL) type 2: ~20% of patients develop pheochromocytoma/paraganglioma. Also causes retinal/cerebellar hemangioblastomas and clear cell renal carcinoma.
- Neurofibromatosis type 1: ~2% develop catecholamine-secreting tumors (usually adrenal pheo).
- Familial paraganglioma (SDHx mutations): Tumors in skull base, neck, thorax, abdomen, or bladder.
Current guidelines recommend genetic screening for all patients with pheochromocytoma, especially with multifocal, bilateral, or metastatic disease.
Morphology
Gross: Ranges from small circumscribed lesions to large hemorrhagic masses weighing several kilograms. Smaller tumors are yellow-tan and well-defined; larger ones tend to be hemorrhagic, necrotic, and cystic. Fresh tissue turns dark brown with potassium dichromate (chromaffin reaction) due to catecholamine oxidation.
Histology: Polygonal to spindle-shaped chromaffin cells arranged in characteristic nests ("Zellballen") separated by a rich vascular network. Cytoplasm is finely granular due to catecholamine-containing granules (highlighted by silver stains). Bizarre cells may be present even in benign tumors.
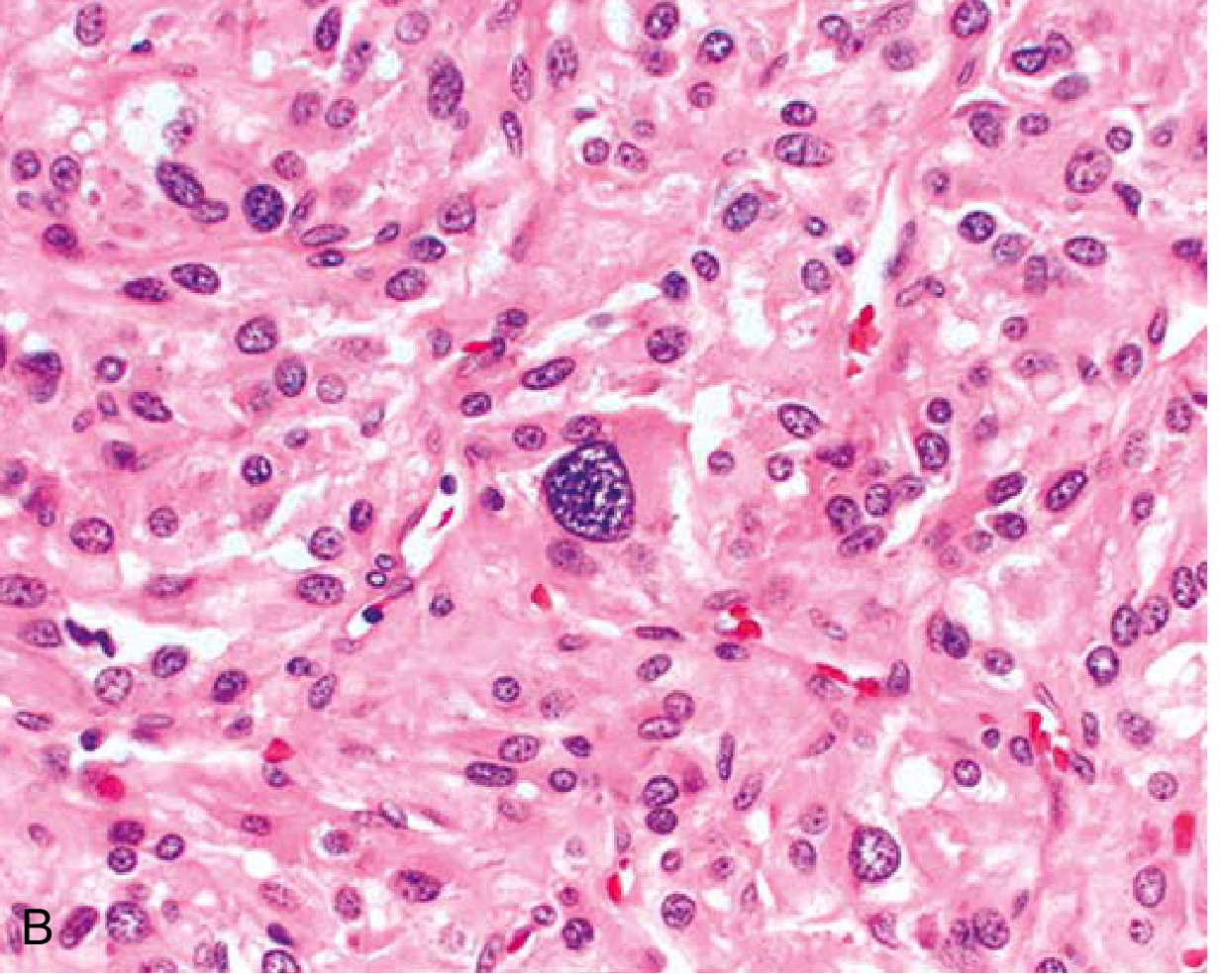
Robbins & Kumar Basic Pathology - Characteristic nests of cells with abundant cytoplasm. Bizarre cells (such as the one in center) can appear even in benign tumors.
Malignancy: Cannot be determined by histology alone - it is defined by the presence of metastases in sites where chromaffin tissue is normally absent (e.g., bone, liver, lymph nodes, lung).
Clinical Features
The dominant clinical manifestation is hypertension, driven by excess catecholamine release:
- Sustained hypertension in most patients
- Paroxysmal hypertensive episodes in ~2/3 of patients - abrupt BP spikes with:
- Tachycardia and palpitations
- Headache (pounding, severe)
- Diaphoresis (profuse sweating)
- Tremor, sense of apprehension
- Abdominal or chest pain, nausea, vomiting
Classic triad: Headache + sweating + hypertension (reported in 95% of cases in large series)
Potentially fatal acute complications during paroxysms:
- Congestive heart failure
- Pulmonary edema
- Myocardial infarction
- Ventricular fibrillation
- Cerebrovascular accidents (stroke)
Other features:
- Some tumors also secrete ACTH (causing Cushing features) or somatostatin
- Normotension is possible - increasingly detected as incidentalomas on imaging
- Bladder paragangliomas cause micturition headache or syncope (catecholamine release during voiding)
Physical clues to associated syndromes:
- Café au lait spots + neurofibromas (NF1)
- Retinal hemangiomas (VHL)
- Port wine stain (Sturge-Weber)
- Ash leaf / shagreen patches + adenoma sebaceum (tuberous sclerosis)
- Marfanoid habitus (MEN2B)
Diagnosis
Step 1 - Biochemical Testing (First)
Imaging is generally not ordered until biochemical evidence of catecholamine excess is obtained.
| Test | Comment |
|---|---|
| Plasma-free metanephrines | Preferred first-line - high sensitivity (~97%), brief snapshot of catecholamine metabolism |
| Urinary fractionated metanephrines | Integrates over 24h, less expensive; more false-negatives in familial/normotensive/intermittently secreting tumors |
| Urinary free catecholamines | Also used |
| Urinary VMA (vanillylmandelic acid) | Traditional marker |
| Clonidine suppression test | For equivocal cases (preferred over glucagon stimulation) |
Key pitfall: False-negative urinary collections are common in familial, normotensive, dopamine-β-hydroxylase-deficient, or intermittently secreting tumors.
Step 2 - Anatomic Imaging
CT scan (preferred for most patients):
- Sensitivity 90-100%
- Pheochromocytoma HU: typically 40-50 on non-contrast CT (>10 HU)
- Vigorous early enhancement with <60% washout in venous phase
- Scan neck/chest/abdomen/pelvis to exclude extraadrenal paraganglioma
MRI:
- Preferred for pediatric patients, pregnant/lactating women, contrast allergy, radiation avoidance
- Pathognomonic: Bright hyperintense signal on T2-weighted images (due to high vascularity and water content)
- Higher sensitivity than CT for extraadrenal paraganglioma (up to 100%)
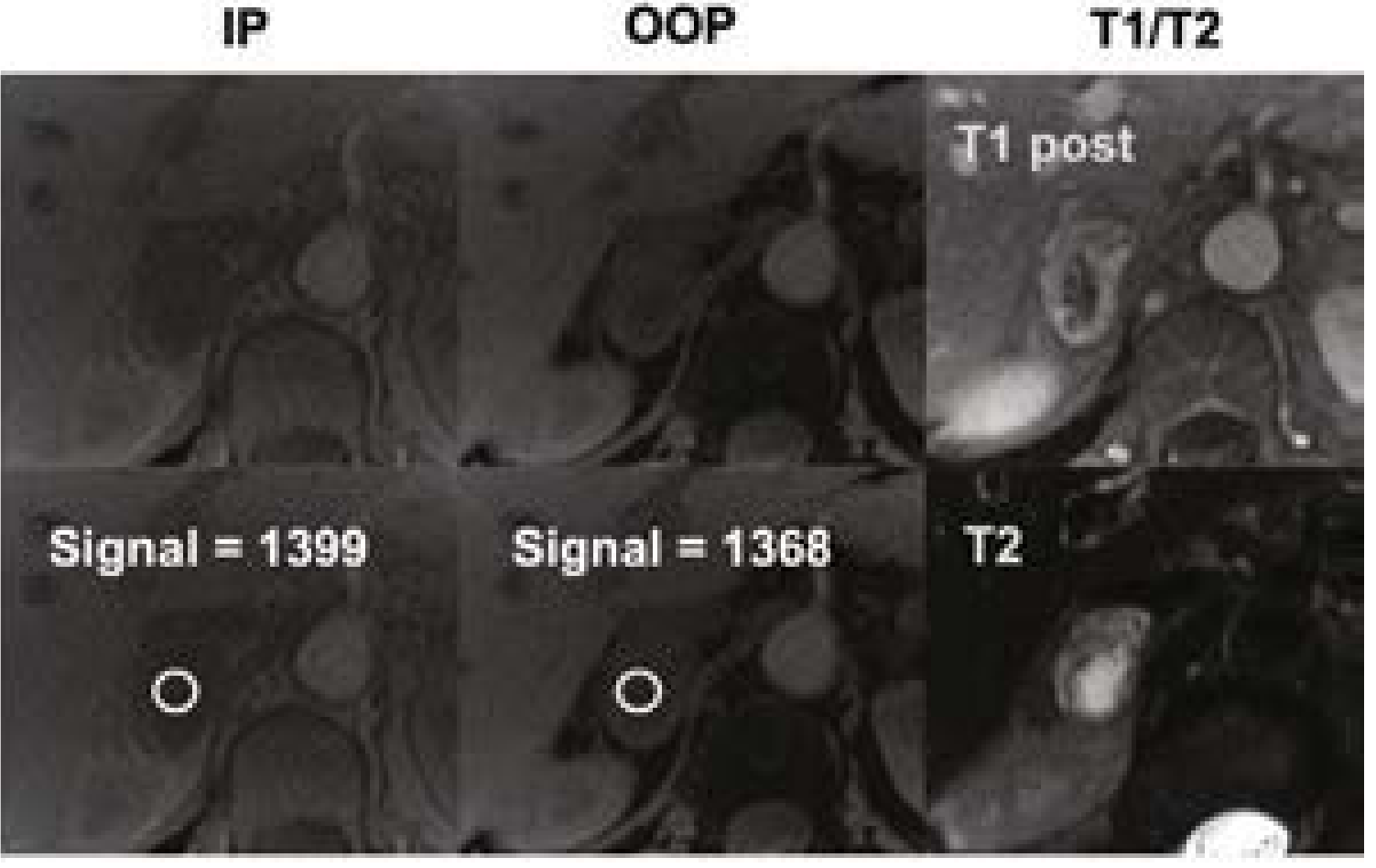
Current Surgical Therapy 14e - MRI showing right adrenal pheochromocytoma: note the T2 hyperintensity (bright signal, lower right), characteristic for pheochromocytomas due to high water content and vascularity.
Step 3 - Functional Imaging (for metastatic/extraadrenal disease)
| Modality | Use |
|---|---|
| ¹²³I-MIBG scintigraphy | Localize/extent of metastatic disease; must stop interfering medications 7-10 days before |
| ¹³¹I-MIBG | Both diagnostic and therapeutic for metastatic disease |
| ¹⁸F-FDG PET/CT | Metabolically active disease |
| ¹⁸F-DOPA PET/CT | Dopaminergic tumors |
| ⁶⁸Ga-DOTATATE PET/CT | Somatostatin receptor-positive tumors |
Management
Pre-operative Pharmacologic Preparation (Critical)
Proper preparation is mandatory before surgery to prevent intraoperative hypertensive crisis:
- Alpha-blockers FIRST - phenoxybenzamine (oral, non-competitive, long-acting) or phentolamine (IV) to block catecholamine effects
- Beta-blockers SECOND (only after alpha-blockade is established) - to control tachycardia. Never give beta-blockers first - unopposed alpha stimulation can cause severe hypertension
- Calcium channel blockers - added before beta-blockers in many protocols
- Wait 7-14 days after localization to normalize BP, heart rate, and intravascular volume (reduces post-resection hypotension)
- Metyrosine (inhibits catecholamine synthesis) - for very large tumors or non-surgical candidates
Surgical Resection
- Laparoscopic adrenalectomy is preferred for most adrenal pheochromocytomas and accessible paragangliomas
- Postoperative monitoring: vigilant monitoring for hypotension, hypoglycemia, adrenal insufficiency
Follow-up
- Repeat biochemical testing 4-6 weeks postoperatively to confirm complete removal
- Long-term annual follow-up, individualized based on pedigree, genetic testing, and recurrence risk factors
- Genetic testing and family screening recommended
Metastatic/Inoperable Disease
- ¹³¹I-MIBG therapy
- Somatostatin analogs for DOTATATE-positive tumors
- Chemotherapy (cyclophosphamide + vincristine + dacarbazine - CVD regimen)
- Long-term alpha-blockade for BP control
Key Distinguishing Points
| Feature | Benign | Malignant |
|---|---|---|
| Histology | Cannot distinguish | Cannot distinguish |
| Diagnosis of malignancy | N/A | Metastases to sites lacking normal chromaffin tissue |
| Extraadrenal location | Less common | Higher malignant rate (up to 20%) |
Sources: Robbins & Kumar Basic Pathology, p. 762-763 | Current Surgical Therapy 14e, p. 870 | Brenner and Rector's The Kidney, p. 2067 | Campbell Walsh Wein Urology
This is a shared conversation. Sign in to Orris to start your own chat.