Congenital cns malformation
Reading File
Finding Sources
Finding Sources
Searching PubMed
"congenital CNS malformations" classification
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have everything needed. Let me compile a thorough, well-structured answer.
Congenital CNS Malformations
Congenital CNS malformations affect approximately 1-2% of pregnancies and are among the leading causes of mental disability and cerebral palsy. Both genetic and environmental factors drive them - genomic sequencing has uncovered many causative variants, while teratogens (chemicals, infections) disrupt normal development at critical windows. Earlier the insult during gestation, the more severe the phenotype.
Classification Overview
CNS malformations fall broadly into:
- Neural Tube Defects (NTDs) - failure of neural tube closure or primary bony defects
- Forebrain Malformations - disorders of proliferation, migration, and organization
- Posterior Fossa Anomalies - cerebellar/brainstem malformations
1. Neural Tube Defects (NTDs)
NTDs are the most common CNS malformations. They are midline defects involving some combination of neural tissue, meninges, and overlying bone/soft tissue.
Two pathogenic mechanisms:
- Failure of neural tube closure (26-28 days gestation) → secondary mesenchymal tissue defects around the malformed tube (e.g., anencephaly, myelomeningocele)
- Primary bony defects from abnormal axial mesoderm development → secondary CNS abnormalities (e.g., encephalocele, meningocele, spina bifida)
Risk factors:
- Folate deficiency in the first trimester (folate supplementation reduces NTD incidence by up to 70%)
- Maternal hyperthermia (e.g., hot tub use)
- Valproate and certain other anticonvulsants
- Multifactorial genetic predisposition
Prenatal screening: Elevated maternal serum alpha-fetoprotein (AFP); AFP also rises in amniotic fluid with open NTDs. Combined with imaging, this allows early detection.
Spina Bifida Spectrum
Spina bifida occulta - Incomplete vertebral arch with no herniation of contents; spinal cord and meninges remain in place. Often asymptomatic, found incidentally.
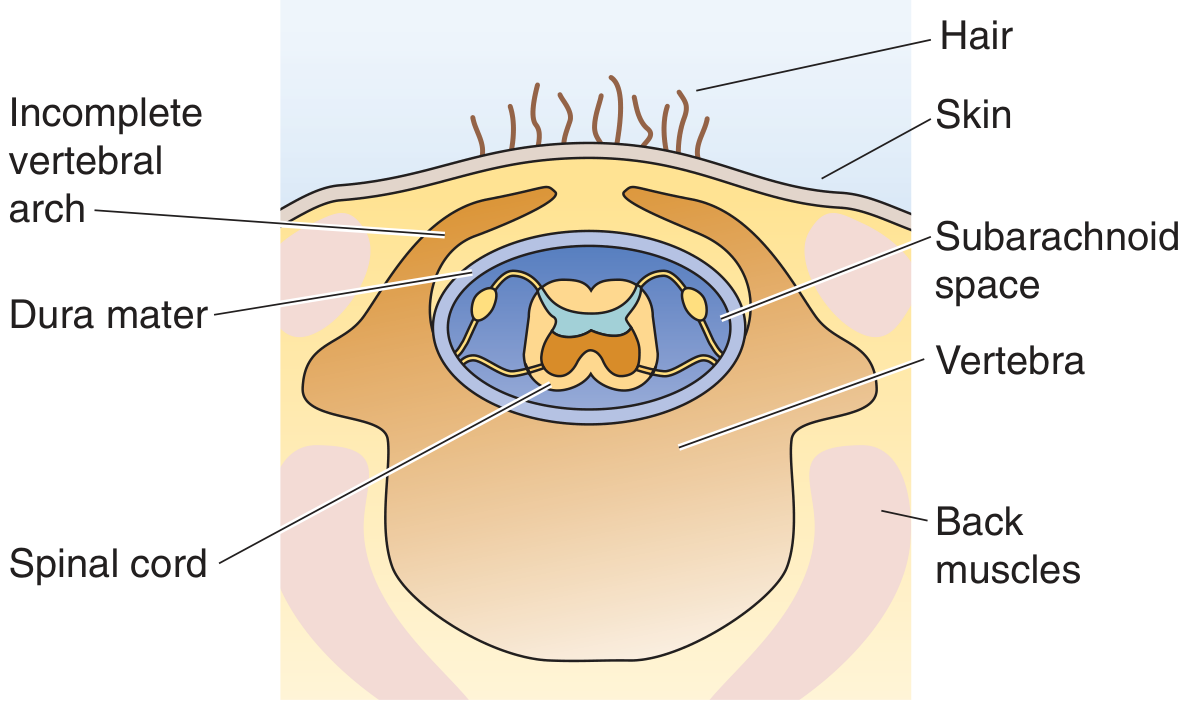
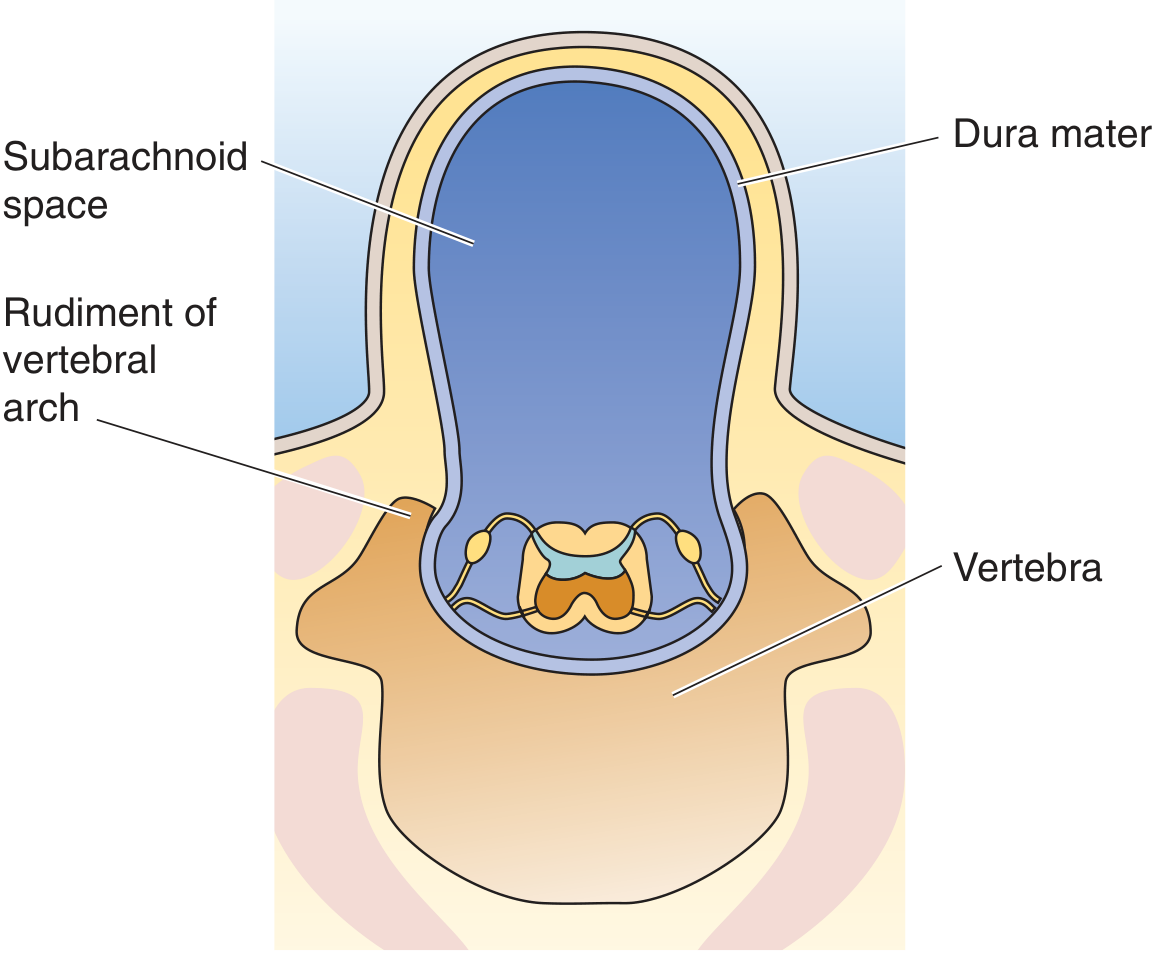
Spina bifida cystica / Meningocele - Dura and arachnoid herniate through a vertebral defect, forming a CSF-filled sac. Spinal cord itself stays in normal position.
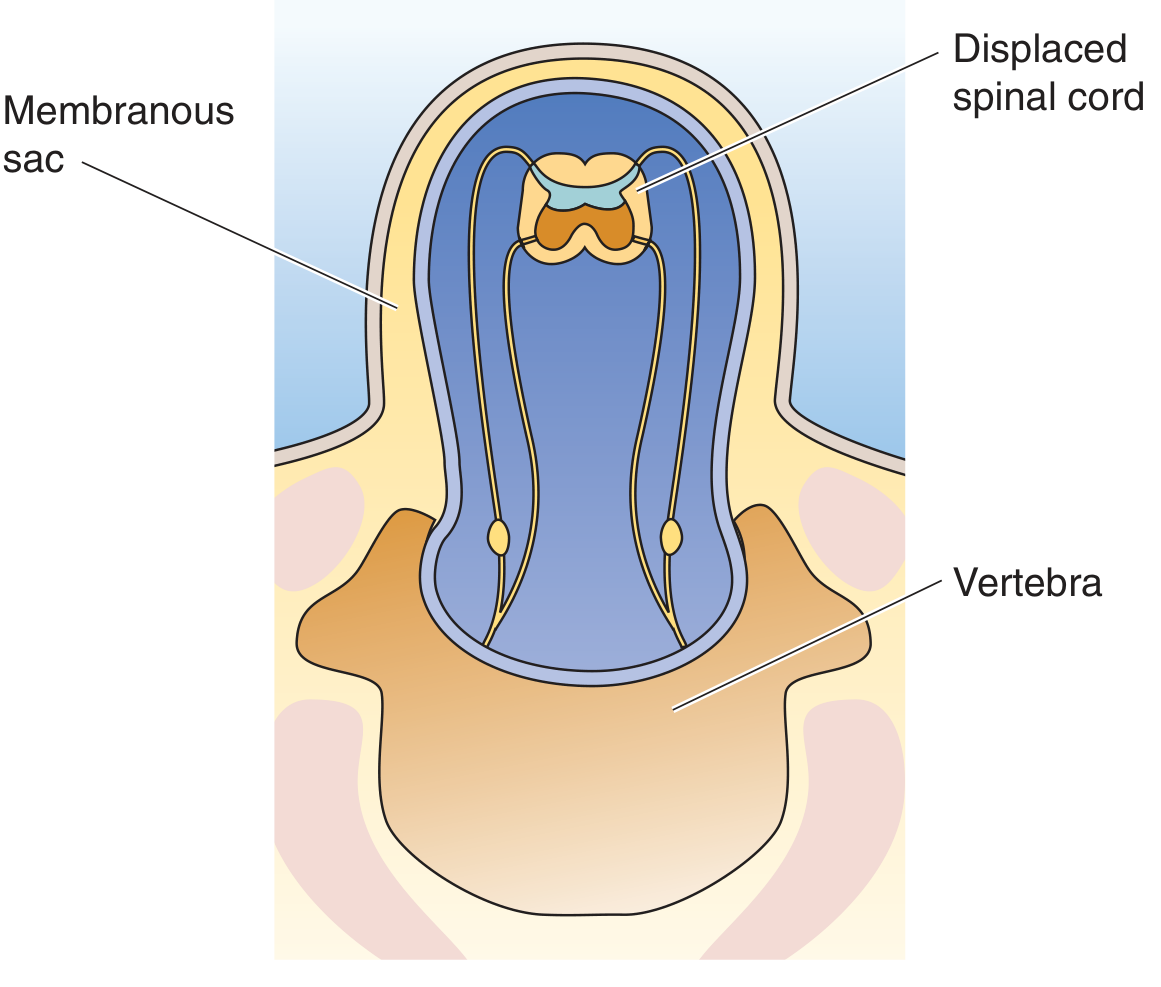
Myelomeningocele (meningomyelocele) - Extension of spinal cord tissue + meninges through a vertebral defect. Most common in the lumbosacral region. Patients have motor/sensory deficits in lower extremities, and bowel/bladder dysfunction. Overlying skin is thin and prone to ulceration/infection.
Variations of spina bifida (Medical Physiology, Fig. 10-7)
Anencephaly
- Malformation of the anterior end of the neural tube
- Forebrain development disrupted at ~28 days gestation
- Cerebral hemispheres absent; calvarium absent
- Remaining tissue = area cerebrovasculosa: disorganized remnant with ependyma, choroid plexus, meningothelial cells
- Posterior fossa structures may be partially spared
- Often leads to spontaneous abortion; incompatible with sustained life
- Occurs in ~1 per 1000 deliveries
Encephalocele
- Diverticulum of malformed CNS tissue extruding through a midline cranial defect
- Most common in the occipital region or posterior fossa
- Anterior variants involve orbit, ethmoid, or cribriform plate ("nasal glial heterotopia" / "nasal glioma")
2. Forebrain Malformations
These result from disruptions of neuronal proliferation, migration, or cortical organization.
Microencephaly (Microcephaly)
- Abnormally small brain volume + small head
- Underlying mechanism: decreased generation of cortical neurons due to premature exit of progenitor cells from the proliferating pool
- Associations: chromosomal abnormalities, fetal alcohol syndrome, HIV, Zika virus infection
Holoprosencephaly
- Failure of normal midline patterning of the prosencephalon
- Mild form (arrhinencephaly): absence of olfactory bulbs and related structures only
- Severe form: brain not divided into hemispheres or lobes; may have facial midline defects including cyclopia
- Genetic cause: loss-of-function mutations in Hedgehog (Shh) signaling pathway
Lissencephaly ("Smooth Brain")
- Loss of gyri - either complete (lissencephaly) or partial (pachygyria)
- Result of failure of normal neuronal migration
- Mutations in migration-controlling genes (e.g., LIS1, DCX/doublecortin)
Polymicrogyria
- Increased number of irregularly formed, small gyri
- Neurons reach the cortex but cortical organization is abnormal
3. Posterior Fossa Anomalies
Arnold-Chiari Malformation (Chiari Type II)
- Small posterior fossa + misshapen midline cerebellum
- Downward extension of the cerebellar vermis through the foramen magnum
- Almost always associated with lumbar myelomeningocele and hydrocephalus
- Clinically significant: causes brainstem compression, syringomyelia
Chiari Type I Malformation
- Far milder than Type II
- Low-lying cerebellar tonsils that descend into the cervical canal (without myelomeningocele)
- May cause headaches, neck pain, syringomyelia
- Often asymptomatic until adulthood
Dandy-Walker Malformation
- Characterized by cystic dilation of the fourth ventricle, agenesis or hypoplasia of the cerebellar vermis, and an enlarged posterior fossa
- Associated with hydrocephalus
4. Syringohydromyelia (Syringomyelia)
- Fluid-filled cavitation within the spinal cord
- Can be a secondary finding in Chiari malformations or after cord injury/inflammation
Diagnostic Summary Table
| Malformation | Key Feature | Association |
|---|---|---|
| Spina bifida occulta | Incomplete vertebral arch only | Often asymptomatic |
| Meningocele | Meningeal herniation, cord intact | Variable |
| Myelomeningocele | Cord + meninges herniation | Motor/sensory deficits, lumbosacral |
| Anencephaly | Absent forebrain + calvarium | Incompatible with life |
| Encephalocele | Brain tissue through skull defect | Occipital most common |
| Holoprosencephaly | Non-divided prosencephalon | Hedgehog mutation, cyclopia |
| Lissencephaly | Absent/reduced gyri | LIS1/DCX mutations |
| Polymicrogyria | Excess, small irregular gyri | Migration defect |
| Chiari II | Vermis herniation + MMC | Hydrocephalus |
| Chiari I | Tonsillar herniation | Syringomyelia |
| Dandy-Walker | 4th ventricle cyst + vermis absence | Hydrocephalus |
Prevention and Detection
- Folic acid supplementation (400-800 mcg/day) periconceptionally reduces NTD risk by up to 70%
- Maternal serum AFP screening at 15-20 weeks: elevated in open NTDs
- Prenatal ultrasound: detects structural anomalies (anencephaly by 11-14 weeks; spina bifida by 18-20 weeks)
- Genetic counseling for families with known mutations (Hedgehog pathway, LIS1, DCX)
Sources: Robbins & Kumar Basic Pathology, p. 822-823; Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 1150; Medical Physiology (Boron & Boulpaep), p. 411-412
This is a shared conversation. Sign in to Orris to start your own chat.