Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism diagram
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive content from multiple authoritative sources. Let me compile the complete answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Overview
ARDS is a clinical syndrome of hypoxemic respiratory failure caused by diffuse alveolar damage (DAD) — an increase in alveolar-capillary permeability that allows protein-rich exudate to flood the airspaces. Unlike cardiogenic pulmonary edema (driven by elevated hydrostatic pressure), ARDS edema is exudative: the alveolar-capillary barrier is disrupted, not passively overwhelmed by pressure. The result is severely impaired gas exchange, reduced lung compliance, refractory hypoxemia, and increased dead-space ventilation.
Triggers and Initiating Insults
ARDS is initiated by either direct (pulmonary) or indirect (extrapulmonary) injury:
| Direct lung injury | Indirect (systemic) injury |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall trigger) |
| Aspiration of gastric contents | Severe trauma / multiple transfusions |
| Inhalation injury | Pancreatitis |
| Near-drowning | Burns |
| Diffuse pulmonary infection | Drug toxicity |
These insults activate the same downstream inflammatory cascade regardless of the entry point.
Three Sequential Pathologic Phases

Figure 312-1 from Harrison's Principles of Internal Medicine 22E — time course of ARDS phases.
Phase 1 — Exudative (Days 0–7)
This is the acute injury phase. Key events:
- Initial insult activates Toll-like receptors (TLRs) on alveolar type I (AT1) epithelial cells and resident alveolar macrophages.
- Macrophages secrete pro-inflammatory cytokines — TNF-α, IL-1β, IL-6, IL-8 — that recruit circulating neutrophils via chemotaxis.
- Neutrophil sequestration: Activated neutrophils become mechanically "stiff" (actin cytoskeleton remodeling) and are physically trapped in narrow pulmonary capillaries (capillary diameter < neutrophil diameter). This produces the transient leukopenia that often precedes ARDS.
- Sequestered neutrophils then transmigrate across the alveolar-capillary membrane, releasing a cascade of cytotoxic mediators:
- Reactive oxygen species (ROS) — oxidative damage to membranes
- Proteases — neutrophil elastase (NE) degrades epithelial/endothelial cadherins (adherens junctions), destroys surfactant protein A, and degrades growth factors
- Neutrophil extracellular traps (NETs) — web-like DNA–histone structures that trap pathogens but also cause endothelial damage and thrombus formation; NET-associated IL-6 and TNF amplify inflammation
- Monocytes migrate into the lung and cause epithelial apoptosis via IFN-β–dependent TRAIL release (activating death receptors on epithelial cells).
- Platelets form aggregates with PMNs and monocytes, amplifying NET formation and inflammatory signaling.
- Endothelial and epithelial barrier failure: Loss of both the microvascular endothelium (necessary and sufficient for edema formation) and the alveolar epithelium leads to flooding of protein-rich fluid and RBCs into the airspaces.
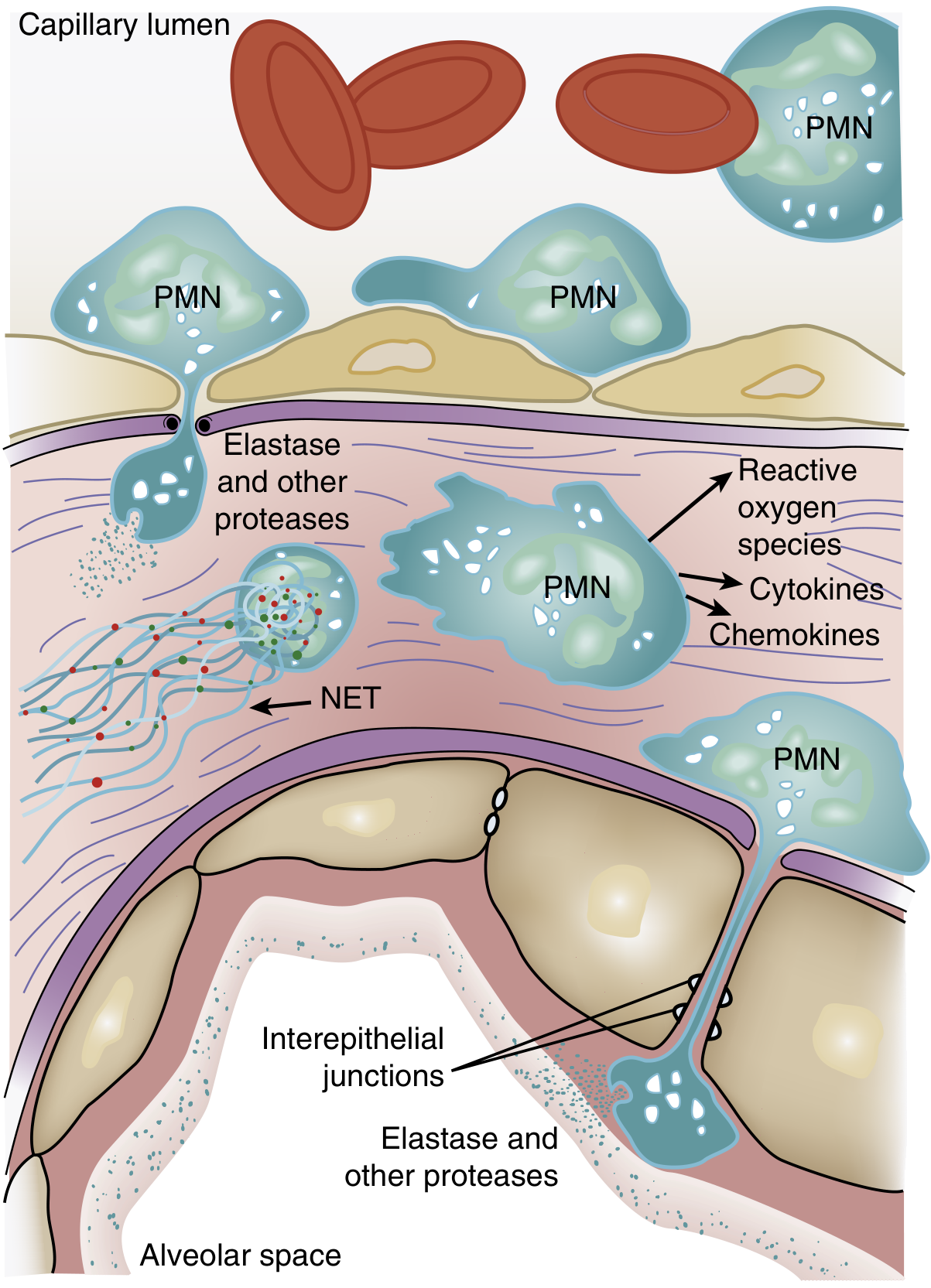
Figure 134.3 from Murray & Nadel's Textbook of Respiratory Medicine — role of neutrophils in ARDS pathogenesis.
Histology of the exudative phase:
- Hyaline membranes (composed of cellular debris, fibrin, and denatured surfactant)
- Alveolar edema (protein-rich fluid)
- Interstitial and alveolar neutrophil infiltration
- Type I pneumocyte necrosis
- Congestion and interstitial edema
Surfactant dysfunction: Leaked plasma proteins (albumin, fibrin) competitively inhibit surfactant. Neutrophil elastase degrades surfactant protein A. The ratio of large (active) to small (inactive) surfactant aggregates falls. The result is alveolar instability and microatelectasis, greatly worsening hypoxemia.
Physiologic consequences:
- Massive right-to-left intrapulmonary shunt (fluid-filled/atelectatic alveoli perfused but not ventilated) → refractory hypoxemia
- Decreased respiratory system compliance (stiff lungs)
- Increased dead-space ventilation (vascular compression, intravascular fibrin deposition)
- Pulmonary hypertension (hypoxic vasoconstriction + fibrin microthrombi + vascular compression by PEEP)
Phase 2 — Proliferative (Days 7–21)
Many patients begin to recover during this phase, but some progress:
- Neutrophil-predominant infiltrate shifts to lymphocyte-predominant
- Type II pneumocytes proliferate along denuded basement membranes, synthesize new surfactant, and transdifferentiate into type I pneumocytes (reepithelialisation)
- Hyaline membranes are reorganized; early fibrotic changes appear (collagen deposition)
- Interstitial inflammation and early architectural disruption develop
- N-terminal procollagen peptide III (a collagen synthesis marker) is detectable in BAL fluid as early as 24 hours after injury onset, indicating fibroproliferation begins simultaneously with inflammation, not after it
Phase 3 — Fibrotic (>21 days, subset of patients)
Not all patients enter this phase, but those who do face serious morbidity:
- Alveolar-duct and interstitial fibrosis replace inflammatory exudate
- Emphysema-like changes with large bullae from architectural destruction
- Intimal fibroproliferation in pulmonary microcirculation → progressive vascular occlusion → pulmonary hypertension
- Marked reduction in lung compliance; increased dead space; risk of pneumothorax
- Lung biopsy evidence of fibrosis in any phase correlates with increased mortality
The Cytokine/Mediator Network
The inflammatory cascade in ARDS is orchestrated by a network of mediators:
| Mediator | Source | Effect |
|---|---|---|
| TNF-α, IL-1β | Macrophages, neutrophils | Amplify inflammation, activate endothelium |
| IL-8 (CXCL8) | Macrophages, epithelium | Key neutrophil chemoattractant |
| IL-6 | Multiple | Acute-phase response; elevated levels = poor prognosis |
| Neutrophil elastase | PMNs | Cleaves cadherins, destroys surfactant, degrades ECM |
| ROS | PMNs, macrophages | Lipid peroxidation, oxidative membrane damage |
| NETs | PMNs | Endothelial injury, thrombus formation, amplify IL-6/TNF |
| Phospholipase A₂ | Pancreas (in pancreatitis) | Degrades surfactant phospholipids |
| p38 MAPK pathway | Inflammatory cells | Stimulates TNF-α production, macrophage chemotaxis |
| TRAIL (via IFN-β) | Monocytes | Epithelial cell apoptosis via death receptor activation |
Coagulation Dysregulation
Intravascular coagulation is a key co-mechanism:
- Tissue factor activation and fibrin deposition in pulmonary microvasculature
- Platelet-neutrophil complexes amplify both coagulation and NET release
- Microthrombi contribute to pulmonary hypertension and dead-space ventilation
- Fibrin components in the alveolar space further impair surfactant function
Impaired Alveolar Fluid Clearance
In normal lungs, alveolar fluid is actively cleared by ENaC (epithelial sodium channels) on type II pneumocytes driving Na⁺ transport, with water following osmotically. In ARDS:
- Hypoxemia and hypercapnia impair sodium transport, reducing fluid clearance
- Loss of type I/II pneumocytes removes the cellular machinery for active clearance
- Edema therefore accumulates and persists, worsening hypoxemia further in a positive-feedback loop
Why DAD Is Found in Only ~50% of ARDS Autopsies
Despite being considered the histologic correlate of ARDS, DAD is confirmed on autopsy or biopsy in only approximately 50% of cases. Patients with confirmed DAD tend to be younger, more severely ill (lower P/F ratios, poorer compliance, higher driving pressures), and are five times more likely to die of hypoxemic respiratory failure. Non-DAD ARDS includes infectious pneumonia, diffuse alveolar hemorrhage, acute eosinophilic pneumonia, and drug pneumonitis — each with distinct and potentially treatable etiologies.
Summary Schematic
Triggering insult (sepsis, pneumonia, aspiration, trauma, etc.)
↓
Activation of TLRs on macrophages and AT1 cells
↓
Release of TNF-α, IL-1β, IL-6, IL-8
↓
Neutrophil recruitment → capillary sequestration → transmigration
↓
PMN degranulation: ROS + elastase + NETs + cytokines
↓
Endothelial + epithelial barrier breakdown
↓
Protein-rich exudate floods alveoli + surfactant dysfunction
↓
Hyaline membrane formation + microatelectasis + shunt
↓
Refractory hypoxemia + ↓ compliance + ↑ dead space + pulm. HTN
↓
(If unresolved) → Type II pneumocyte proliferation → fibroproliferation
↓
(Subset) → Fibrotic phase: pulmonary fibrosis + bullae + cor pulmonale
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134 (Etiology and Pathogenesis of ARDS)
- Harrison's Principles of Internal Medicine, 22nd Ed., Chapter 312 (Clinical Course and Pathophysiology)
- Robbins & Kumar Basic Pathology (Acute Lung Injury and ARDS)
- Goldman-Cecil Medicine, Chapter 90–91 (ARDS and Mechanical Ventilation)
This is a shared conversation. Sign in to Orris to start your own chat.