Explain in detail pharmacokinetics
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have all the material needed for a detailed, well-sourced answer. Here is the full explanation:
Pharmacokinetics: A Detailed Overview
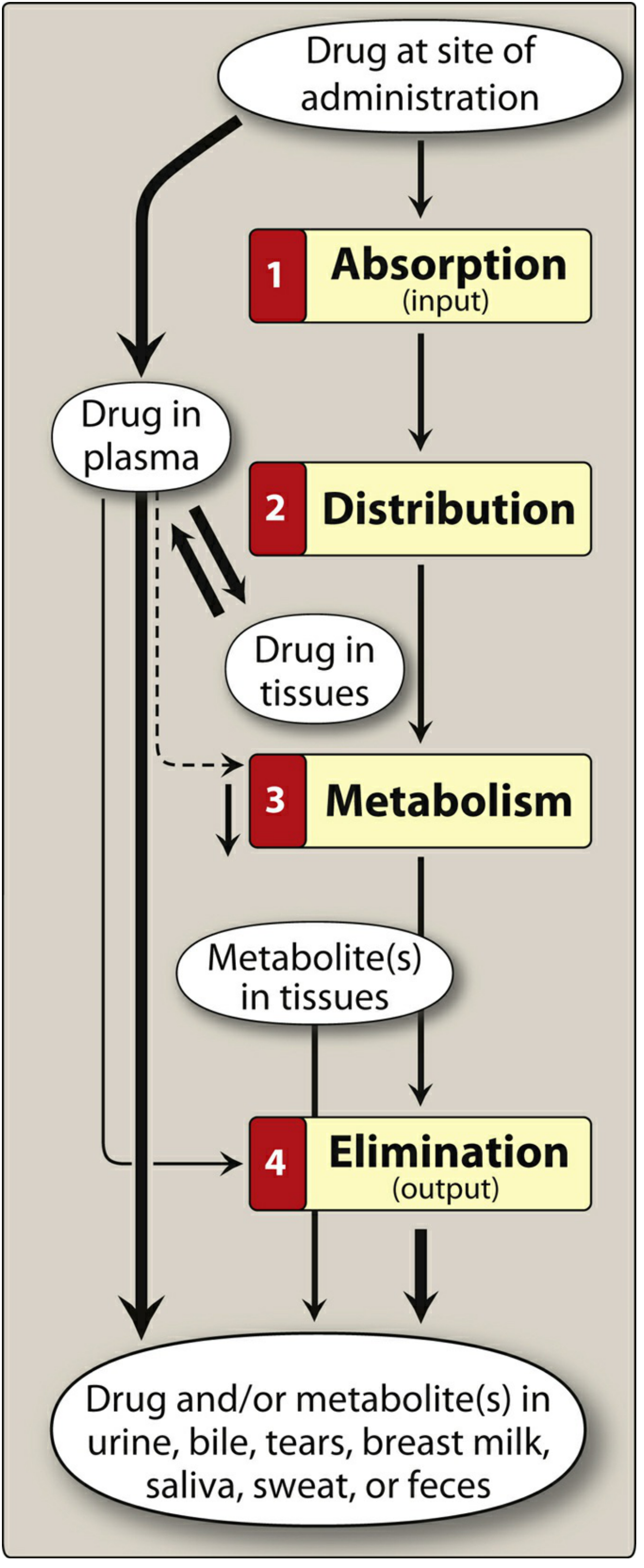
Figure: The ADME cycle. - Lippincott Illustrated Reviews: Pharmacology, p. 20
Pharmacokinetics describes what the body does to a drug. It encompasses four sequential processes - Absorption, Distribution, Metabolism, and Elimination (ADME) - that collectively determine the onset, intensity, and duration of drug action. In contrast, pharmacodynamics describes what the drug does to the body. Understanding PK allows clinicians to design optimal drug regimens (dose, route, frequency, and duration).
1. Absorption
Absorption is the process by which a drug moves from its site of administration into the systemic circulation (plasma).
Routes of Administration
The route chosen depends on the drug's physicochemical properties (lipid/water solubility, ionization) and the therapeutic goal (rapid onset, local effect, sustained release).
| Route | Absorption | Advantages | Disadvantages |
|---|---|---|---|
| Oral | Variable; affected by pH, food, GI motility | Most convenient and economical | Subject to first-pass metabolism; gastric acid may degrade drug |
| Sublingual / Buccal | Rapid direct systemic absorption for select drugs | Bypasses first-pass effect; avoids gastric acid | Limited to small doses; few eligible drugs |
| Intravenous (IV) | No absorption step required | Immediate effect; 100% bioavailability | Risk of infection, embolism; irreversible |
| Intramuscular (IM) | Rapid (aqueous) or slow (depot) | Depot formulations possible | Painful; must avoid certain vessels |
| Subcutaneous (SC) | Slower than IM | Good for insulin, vaccines | Limited volume |
| Transdermal | Slow, variable by skin site | Avoids first-pass; sustained delivery | Only lipophilic drugs; skin irritation possible |
| Rectal | Often erratic and incomplete | Bypasses ~50% of portal circulation; useful if vomiting | Incomplete and unpredictable; rectal irritation |
| Inhalation | Rapid; large surface area | Direct lung delivery; minimizes systemic effects | Requires coordination (MDIs); irritation |
| Intrathecal | Direct CNS delivery | Bypasses blood-brain barrier (BBB) | Invasive; risk of infection |
(Lippincott Illustrated Reviews: Pharmacology, p. 21-30)
Key Concepts in Absorption
Bioavailability (F): The fraction of an administered dose that reaches the systemic circulation unchanged. IV administration gives F = 1 (100%). Oral bioavailability is reduced by incomplete absorption and first-pass metabolism.
First-pass effect (presystemic metabolism): Orally administered drugs are absorbed from the GI tract and travel via the portal vein to the liver before reaching systemic circulation. The liver can extensively metabolize some drugs, markedly reducing their effective dose. Examples: nitroglycerin (virtually completely destroyed orally - hence sublingual use), lidocaine, propranolol, morphine. Sublingual and rectal routes partially bypass this effect.
Factors affecting oral absorption:
- Gastric pH: Low pH inactivates acid-labile drugs (e.g., penicillin G). Enteric-coated preparations resist dissolution until they reach the higher-pH small intestine.
- GI motility: Faster motility reduces contact time, decreasing absorption; slower motility increases it.
- Drug formulation: Enteric-coated tablets, extended-release capsules, and prodrugs are designed to control the timing and site of drug release.
- Ionization (pKa and Henderson-Hasselbalch equation): Only unionized (non-polar) drug can cross lipid membranes. Weak acids (e.g., aspirin) are better absorbed in the acidic stomach; weak bases (e.g., most drugs) are better absorbed in the slightly alkaline small intestine.
2. Distribution
After absorption, the drug enters plasma and then distributes into interstitial and intracellular fluids and tissues.
Protein Binding
Most drugs bind reversibly to plasma proteins, primarily albumin (for acidic drugs) and alpha-1-acid glycoprotein (for basic drugs). Only the free (unbound) fraction is pharmacologically active and available for metabolism and elimination. Bound drug acts as a reservoir - as free drug is eliminated, bound drug dissociates to replenish it, maintaining a relatively constant free-drug fraction.
- Drugs can also bind to tissue proteins, lipids, and nucleic acids, leading to tissue concentrations that far exceed plasma concentrations. This can cause local toxicity (e.g., acrolein accumulates in the bladder causing hemorrhagic cystitis).
Lipophilicity
Lipophilic drugs readily cross cell membranes and distribute widely throughout tissues. The main determinant of their distribution is blood flow to the area. Hydrophilic drugs are largely restricted to extracellular fluid because they cannot cross lipid bilayers.
Volume of Distribution (Vd)
$$V_d = \frac{\text{Amount of drug in the body}}{C_0}$$
Vd is a hypothetical volume that would be required to contain all the drug at the concentration found in plasma. It has no direct physiological meaning but is clinically useful:
| Vd | Location | Example |
|---|---|---|
| ~4 L (plasma volume) | Drug stays in plasma (large MW or highly protein-bound) | Heparin |
| ~14 L (extracellular fluid) | Drug is hydrophilic; crosses capillaries but not cells | Aminoglycosides |
| ~42 L (total body water) | Drug is lipophilic; crosses all membranes freely | Ethanol, chloroquine |
| >100 L | Extensive tissue binding; very little in plasma | Amiodarone, digoxin |
A large Vd indicates extensive tissue distribution; a small Vd indicates confinement to plasma or extracellular fluid. (Lippincott Illustrated Reviews: Pharmacology, p. 48)
Special Distribution Barrier: Blood-Brain Barrier (BBB)
The BBB is formed by tight junctions between brain capillary endothelial cells. Only lipophilic, unionized drugs can cross it. Highly ionized or protein-bound drugs are excluded. P-glycoprotein (Pgp/MDR1), an ABC transporter in the BBB, actively pumps many drugs back out, limiting CNS penetration and conferring drug resistance. (Goodman & Gilman's, p. 99)
Membrane Transporters
The SLC (solute carrier) and ABC transporter superfamilies are critical in determining drug distribution. Transporters in intestinal, renal, and hepatic epithelia govern selective absorption and elimination. Pgp, OAT, OCT, and OATP are clinically relevant examples that explain why some drugs fail to reach target tissues or accumulate unexpectedly. (Goodman & Gilman's, p. 332)
3. Metabolism (Biotransformation)
The liver is the primary site of drug metabolism, though the gut wall, lungs, plasma, and kidneys also contribute. The goal of metabolism is generally to convert lipophilic drugs into more polar (hydrophilic) metabolites that can be excreted by the kidneys or bile. Metabolites are usually inactive, but some are active (prodrugs) or even toxic.
Phase I Reactions - "Functionalization"
These reactions introduce or unmask a polar functional group (-OH, -NH2, -SH, -COOH), making the drug slightly more water-soluble. The product may be pharmacologically active, inactive, or toxic.
Cytochrome P450 (CYP) Enzymes are the most important Phase I enzymes. They are located primarily in hepatocyte endoplasmic reticulum. Key isozymes include:
| CYP Isozyme | Major Substrates | Clinical Importance |
|---|---|---|
| CYP3A4 | ~50% of all drugs (statins, calcium-channel blockers, macrolides, cyclosporine) | Most abundant hepatic CYP; major target for drug interactions |
| CYP2D6 | Beta-blockers, codeine, antidepressants (SSRIs) | Genetic polymorphism - poor vs. extensive metabolizers |
| CYP2C9 | Warfarin, NSAIDs, phenytoin | Narrow therapeutic index drugs - major interaction site |
| CYP2C19 | Omeprazole, clopidogrel | Polymorphism affects clopidogrel activation |
| CYP1A2 | Caffeine, theophylline, clozapine | Induced by smoking |
CYP Inducers (e.g., rifampin, phenytoin, carbamazepine, St. John's Wort) upregulate CYP expression, increasing drug metabolism and reducing plasma drug levels - this can cause therapeutic failure.
CYP Inhibitors (e.g., ketoconazole, clarithromycin, ritonavir, omeprazole) competitively or non-competitively block CYP, decreasing drug metabolism and elevating plasma drug levels - this can cause toxicity. For example, omeprazole inhibits three CYP isozymes that metabolize warfarin, increasing anticoagulant effect and bleeding risk. (Lippincott Illustrated Reviews: Pharmacology, p. 502)
Other Phase I reactions (non-P450):
- Monoamine oxidase (MAO) - oxidizes catecholamines, histamine
- Alcohol dehydrogenase - metabolizes ethanol
- Esterases - hydrolyze aspirin in the liver
- Hydrolysis - e.g., methylphenidate
Phase II Reactions - "Conjugation"
If Phase I products are still not polar enough for excretion, they undergo conjugation with endogenous molecules to form highly polar, usually inactive conjugates for renal or biliary excretion.
| Conjugation Reaction | Endogenous Substrate | Example |
|---|---|---|
| Glucuronidation (most common) | UDP-glucuronic acid | Morphine (morphine-6-glucuronide is MORE potent) |
| Sulfation | Sulfuric acid | Acetaminophen, steroids |
| Acetylation | Acetyl-CoA | Isoniazid, sulfonamides |
| Methylation | SAM | Norepinephrine, histamine |
| Amino acid conjugation | Glycine, glutamine | Salicylic acid |
| Glutathione conjugation | Glutathione | NAPQI (toxic acetaminophen metabolite) |
Some drugs with pre-existing -OH, -NH2, or -COOH groups bypass Phase I entirely and go directly to Phase II. (Lippincott Illustrated Reviews: Pharmacology, p. 506)
First-Order vs. Zero-Order Kinetics
- First-order kinetics: A constant fraction of drug is metabolized per unit time. Most drugs follow this. As drug concentration falls, so does the rate of metabolism. This gives a linear log-concentration vs. time plot.
- Zero-order kinetics: A constant amount of drug is metabolized per unit time regardless of concentration (enzyme saturation). Examples: ethanol, phenytoin, aspirin at toxic doses. This is dangerous because small dose increases produce disproportionately large rises in plasma concentration.
4. Elimination (Excretion)
The kidney is the principal organ of excretion. Drugs must be sufficiently polar (or converted to polar metabolites) to be eliminated. Other routes include bile/feces, lungs (volatile anesthetics), breast milk, saliva, and sweat.
Renal Elimination - Three Processes
1. Glomerular Filtration
Free (unbound) drug is filtered passively at the glomerulus at a rate proportional to the GFR (~120 mL/min in a healthy adult). Protein-bound drug is too large to pass through the capillary slits. Lipid solubility and pH do not affect filtration. Reduction in GFR (renal disease, aging) directly impairs drug elimination.
2. Active Tubular Secretion
Drugs that were not filtered (because they are protein-bound) are secreted by active transport into the proximal tubule. There are separate transporter systems for organic anions (OAT) and organic cations (OCT). Because transporters can be saturated, competition between drugs for the same transporter can occur - e.g., probenecid blocks penicillin secretion, prolonging its action.
3. Passive Tubular Reabsorption
As the glomerular filtrate is concentrated in the tubules, lipophilic drugs diffuse back across the tubular epithelium into plasma. Highly ionized drugs cannot be reabsorbed and are excreted. This is the basis for urinary pH manipulation to treat overdoses:
- Alkalinizing urine (sodium bicarbonate) traps weak acids (e.g., aspirin, phenobarbital) in ionized form - preventing reabsorption and increasing excretion.
- Acidifying urine (ammonium chloride) traps weak bases (e.g., amphetamines) - similarly enhancing their excretion.
(Lippincott Illustrated Reviews: Pharmacology, p. 513-522)
5. Key PK Parameters
Clearance (CL)
Clearance is the volume of plasma from which drug is completely removed per unit time. Total body clearance is the sum of hepatic clearance, renal clearance, and other clearances. It determines the maintenance dose rate needed to achieve a target steady-state concentration.
$$CL = \frac{\text{Rate of elimination}}{\text{Plasma drug concentration}}$$
Half-Life (t½)
The time required for plasma drug concentration to fall by 50%. For first-order kinetics:
$$t_{1/2} = \frac{0.693 \times V_d}{CL}$$
- t½ is inversely proportional to clearance and directly proportional to Vd.
- It takes approximately 4-5 half-lives to reach steady-state concentration during repeated dosing.
- It also takes 4-5 half-lives to essentially eliminate a drug.
Bioavailability (F) and AUC
The area under the plasma concentration-time curve (AUC) represents total drug exposure. Bioavailability is calculated by comparing the AUC after a route of administration to the AUC after IV administration:
$$F = \frac{AUC_{oral}}{AUC_{IV}} \times 100%$$
Steady State (Css)
With repeated dosing, the rate of drug input equals the rate of elimination. Steady-state is reached after 4-5 half-lives. The average steady-state concentration depends on the dose, dosing interval, bioavailability, and clearance.
Loading dose: Used when a rapid effect is needed. Calculated as:
$$\text{Loading dose} = V_d \times C_{target}$$
Maintenance dose: Sustains steady-state concentration:
$$\text{Maintenance dose} = CL \times C_{ss} \times \tau$$
(where τ = dosing interval)
6. Special Considerations
Hepatic Impairment
Reduces Phase I and Phase II metabolism. Drugs with high first-pass extraction (high hepatic extraction ratio) are most affected - their bioavailability increases dramatically in liver disease (e.g., propranolol, morphine). Protein synthesis also falls, reducing albumin and increasing free drug fraction.
Renal Impairment
Reduces glomerular filtration, tubular secretion, and tubular reabsorption. Renally cleared drugs (aminoglycosides, digoxin, vancomycin, metformin) accumulate and require dose reduction or interval extension. Creatinine clearance (CrCl) is used to estimate remaining renal function.
Aging
Older adults have reduced GFR, reduced hepatic blood flow and CYP activity, decreased albumin, and increased body fat - all of which alter PK. Drug half-lives are prolonged and toxicity risk is higher.
Pregnancy
Physiological changes (increased plasma volume, increased GFR, altered protein binding, induced CYP enzymes) can significantly alter PK. Some drugs cross the placenta and affect the fetus (teratogenicity risk).
Pharmacogenetics
Genetic polymorphisms in CYP2D6, CYP2C19, CYP2C9, and drug transporters create inter-individual variability in drug response. "Poor metabolizers" lack functional enzyme (drug accumulates - toxicity risk); "ultrarapid metabolizers" have extra gene copies (drug cleared too quickly - therapeutic failure). This is the basis for pharmacogenomic testing before prescribing drugs like clopidogrel, codeine, or warfarin.
Summary Table
| Parameter | Definition | Clinical Use |
|---|---|---|
| Bioavailability (F) | Fraction reaching systemic circulation | Determines oral dose vs. IV dose |
| Volume of Distribution (Vd) | Apparent fluid volume containing all drug | Estimates loading dose; predicts dialyzability |
| Clearance (CL) | Volume of plasma cleared per unit time | Determines maintenance dose |
| Half-life (t½) | Time for 50% reduction in plasma concentration | Determines dosing frequency; time to steady state |
| AUC | Total drug exposure over time | Measures bioavailability and overall exposure |
| Steady-State (Css) | Plateau concentration with repeated dosing | Target for therapeutic drug monitoring |
| First-pass effect | Hepatic pre-systemic metabolism | Explains oral dose adjustments |
Sources:
- Lippincott Illustrated Reviews: Pharmacology, Chapter 1 (Pharmacokinetics), pp. 19-70
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, Chapters 2 & 4 (ADME & Drug Transport)
This is a shared conversation. Sign in to Orris to start your own chat.