Pathophysiology of allergic rhinitis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"allergic rhinitis"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive material from multiple authoritative textbooks. Let me compose the full detailed answer.
Pathophysiology of Allergic Rhinitis
Allergic rhinitis (AR) is the most common manifestation of immunopathology in the upper aerodigestive tract, affecting 10-20% of people in the United States and Europe. Its pathophysiology is fundamentally a Type I (IgE-mediated) hypersensitivity reaction driven by Th2-skewed adaptive immunity, with mast cell degranulation as the central early event and a chronic eosinophil/lymphocyte-dominated inflammatory response sustaining nasal hyperresponsiveness.
1. Sensitization Phase (First Exposure)
When an allergen (pollen, dust mite, pet dander, fungal spore) is first inhaled, it is processed by dendritic cells in the nasal mucosa. These antigen-presenting cells migrate to regional lymph nodes and present allergen peptides on MHC class II molecules to naive CD4+ T cells.
Under atopic conditions, the immune microenvironment favors differentiation of these T cells into the Th2 phenotype (driven by IL-4). Th2 cells produce a characteristic cytokine profile:
| Cytokine | Key Effect |
|---|---|
| IL-4 | Drives B-cell class switching to IgE; promotes Th2 differentiation |
| IL-5 | Eosinophil proliferation, activation, and survival |
| IL-13 | Mucus hypersecretion, epithelial barrier disruption |
| IL-9 | Mast cell growth and function |
Under the influence of IL-4 and IL-13, B cells undergo class switching and produce allergen-specific IgE antibodies. These IgE molecules circulate and bind with high affinity to FcεRI receptors on the surface of tissue mast cells and peripheral basophils, "arming" them for rapid response upon re-exposure.
2. Early Phase Response (Minutes After Re-Exposure)
On subsequent allergen exposure, the inhaled antigen cross-links adjacent IgE molecules on the surface of sensitized mast cells. This IgE cross-linking triggers a rapid intracellular signaling cascade via Syk kinase and downstream phospholipase C activation, resulting in:
Preformed Mediators Released by Degranulation:
- Histamine - the dominant mediator; binds H1 receptors on blood vessels and sensory nerves causing vasodilation, increased vascular permeability, glandular hypersecretion, and activation of sensory nerve endings (sneezing, itch)
- Tryptase - a serine protease; marker of mast cell activation; contributes to tissue remodeling
- Kinins (bradykinin) - vasodilation, pain, increased vascular permeability
Newly Synthesized Mediators (Minutes):
- Prostaglandin D2 (PGD2) - vasodilation, mucus secretion, nasal congestion
- Leukotrienes C4, D4, E4 (cysteinyl leukotrienes) - potent vasodilators and bronchoconstrictors; major contributors to nasal congestion; 100-1,000x more potent than histamine on a molar basis
- Platelet-activating factor (PAF) - recruits eosinophils, increases vascular permeability
Clinical result: Within minutes - sneezing, profuse watery rhinorrhea, nasal pruritus, and conjunctivitis (from sensory nerve stimulation and vasodilation). Nasal congestion from tissue edema begins in the early phase but dominates the late phase.
3. Late Phase Response (4-8 Hours After Exposure)
Chemoattractants and adhesion molecules released during the early phase (including eotaxin, RANTES/CCL5, IL-5, and ICAM-1) promote recruitment of a second wave of inflammatory cells to the nasal mucosa:
- Eosinophils - the hallmark late-phase cell; release major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil peroxidase, and leukotrienes; cause epithelial damage and perpetuate inflammation
- Basophils - produce histamine and IL-4 upon activation; amplify the Th2 response
- CD4+ Th2 lymphocytes - sustain cytokine production (IL-4, IL-5, IL-13)
- Monocytes - differentiate into macrophages and contribute to chronic inflammation
Clinical result: Nasal congestion is the dominant symptom (4-10 hours after exposure, peaking around 6 hours, resolving by 24 hours). Elevated nasal airway resistance reflects mucosal edema from this cellular infiltration. Post-nasal drip and persistent malaise are also characteristic.
During the late-phase, cytokine levels in nasal lavage rise markedly: IL-1β, TNF-α, GM-CSF in the early hours; then IL-5, IL-6, IL-8, GM-CSF, and soluble ICAM-1 in the late phase.
4. Nasal Mucosal Hyperresponsiveness and Priming
Repeated allergen exposure leads to amplification of mucosal hyperresponsiveness - a phenomenon called "priming." The nasal mucosa becomes increasingly reactive to both specific allergens and nonspecific irritants (cold air, strong odors, smoke). This explains why in seasonal AR, symptom severity depends not just on the current pollen count but on cumulative allergen exposure for the entire season - severe symptoms can develop at pollen levels that would have been tolerated early in the season.
Priming involves:
- Upregulation of FcεRI receptor expression on mast cells
- Increased sensitivity of sensory nerve endings (neuropeptide-mediated neurogenic inflammation)
- Ongoing cytokine production maintaining a pro-inflammatory milieu in the submucosa
5. Neurogenic Amplification
The nasal mucosa is richly innervated by unmyelinated nociceptive C fibers that release neuropeptides amplifying the allergic response:
- Tachykinins (substance P, neurokinin A) - vasodilation, plasma extravasation, mucus secretion
- Calcitonin gene-related peptide (CGRP) - potent vasodilator
- Vasoactive intestinal peptide (VIP) from parasympathetic endings - nasal secretion, vasodilation
- Neuropeptide Y from sympathetic endings - vasoconstriction (counterregulatory)
This "neurogenic inflammation" also explains the nasal-bronchial reflex - stimulation of nasal sensory nerves can trigger bronchospasm, contributing to the well-recognized co-occurrence of AR with asthma (about 80% of patients with allergic asthma also have AR).
6. Chronic Inflammation and Structural Changes
In persistent AR, chronic mucosal inflammation leads to:
- Subepithelial fibrosis and basement membrane thickening
- Goblet cell hyperplasia and mucus gland hypertrophy - contributing to chronic rhinorrhea
- Epithelial barrier disruption - increased permeability facilitates further allergen penetration
- Nasal polyp formation - recurrent episodes can cause focal mucosal protrusions; histologically characterized by edematous stroma, cystic mucus glands, and eosinophil-rich infiltrates
7. Histopathology
The nasal mucosa in AR shows:
- Mucosal edema and erythema
- Prominent eosinophilic infiltrate in the lamina propria
- Increased numbers of mast cells in the epithelium and submucosa
- Goblet cell hyperplasia
- Increased CD4+ T cells and IgE-bearing B cells
- Thickened subepithelial basement membrane
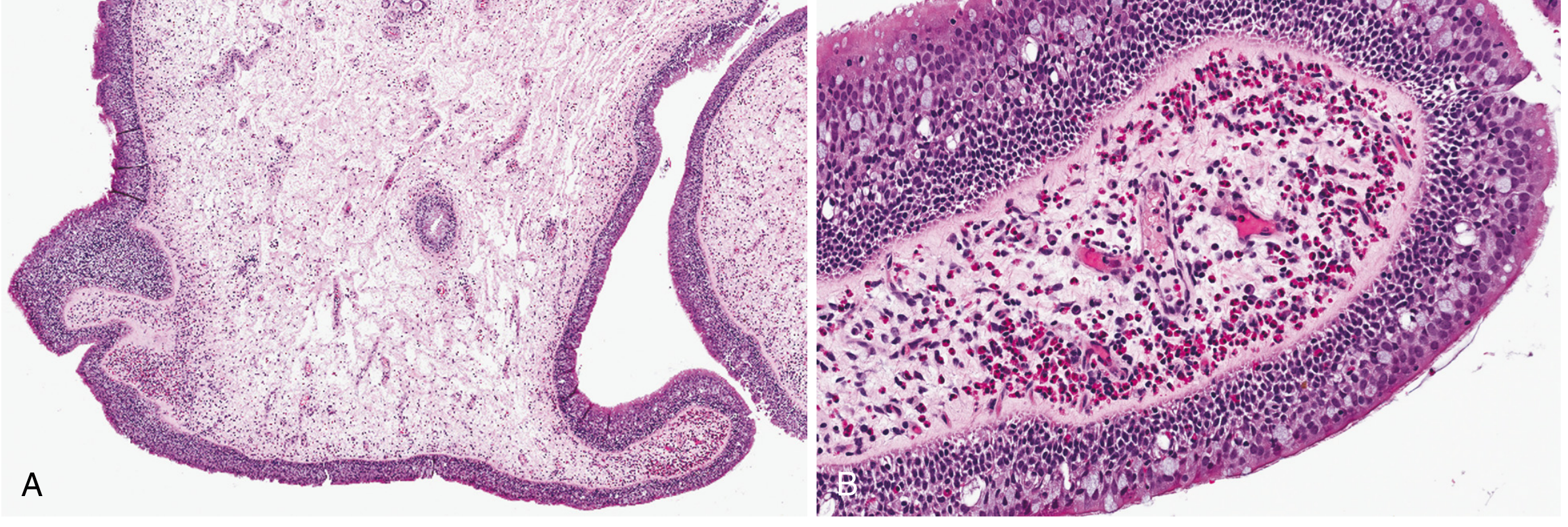
Histology of nasal polyp complicating allergic rhinitis - Robbins Pathology. Note the dense eosinophilic infiltrate (pink-red granular cells) surrounding dilated vessels in the edematous stroma.
8. Unified Airway Disease
AR and asthma share a common pathophysiology (Th2-driven, IgE-mediated) and strong epidemiologic links. The concept of "unified airway disease" reflects that the upper and lower respiratory tracts should be considered as one functional unit. Nasal inflammation can directly contribute to lower airway disease through:
- Mouth breathing bypassing nasal filtration and humidification
- Post-nasal drip triggering bronchial irritation
- Systemic allergic inflammation reaching the bronchial circulation
Summary Diagram
| Phase | Timing | Key Cells | Key Mediators | Symptoms |
|---|---|---|---|---|
| Sensitization | First exposure | Dendritic cells, Th2, B cells | IL-4, IL-13, IgE | None (silent) |
| Early | 0-60 min | Mast cells, basophils | Histamine, leukotrienes, PGD2 | Sneezing, rhinorrhea, itch |
| Late | 4-8 hrs | Eosinophils, T cells, basophils | IL-5, ECP, MBP, leukotrienes | Congestion, malaise |
| Chronic | Persistent | All above + fibroblasts | TGF-β, IL-13, remodeling factors | Persistent obstruction, polyps |
Sources:
- Cummings Otolaryngology Head and Neck Surgery, Chapter 35
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter on Allergic Rhinitis
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.