Glomerular nephritis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"glomerulonephritis"[MeSH Terms] AND "diagnosis"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive content. Let me compile the full, detailed response.
Glomerulonephritis (GN) - Comprehensive Overview
Definition
Glomerulonephritis refers to a group of kidney diseases characterized by inflammation of the glomeruli, leading to disruption of the normal filtration barrier. It is the third most common cause of end-stage kidney disease (ESKD) worldwide. The diagnosis requires serum chemistry, serology, urinalysis with microscopy, and quantification of proteinuria - and often kidney biopsy for definitive classification.
- National Kidneys Foundation Primer on Kidney Diseases, 8e, p. 192
- Comprehensive Clinical Nephrology, 7th Edition
Clinical Syndromes
GN presents as distinct clinicopathological syndromes. Understanding which syndrome is present is the key first step in narrowing the differential:
1. Nephrotic Syndrome
- Proteinuria >3.5 g/24 hr, serum albumin <3.5 g/dL, hyperlipidemia, edema
- Onset is insidious; heavy pitting edema, normal blood pressure (initially)
- No red cell casts; serum albumin markedly low
- Complications: hypercoagulability (loss of protein C, S, antithrombin III), renal vein thrombosis (risk when proteinuria >10 g/24 hr + albumin <2 g/dL), iron deficiency anemia (loss of transferrin), vitamin D deficiency, and hypogammaglobulinemia
- Common causes: Minimal Change Disease (MCD), FSGS, Membranous Nephropathy, Diabetic Nephropathy, Amyloidosis
2. Nephritic Syndrome
- Glomerular hematuria (dysmorphic RBCs, RBC casts), hypertension, oliguria, elevated creatinine, non-nephrotic proteinuria
- Onset is abrupt; raised jugular venous pressure, raised blood pressure
- Reflects active glomerular inflammation with proliferation and leukocyte infiltration
- Reduced GFR from capillary wall injury; hypertension from fluid retention + renin release
| Feature | Nephrotic | Nephritic |
|---|---|---|
| Onset | Insidious | Abrupt |
| Edema | ++++ | ++ |
| Blood pressure | Normal | Raised |
| Proteinuria | ++++ | ++ |
| Hematuria | May/may not occur | +++ |
| RBC casts | Absent | Present |
| Serum albumin | Low | Normal/slightly reduced |
- Comprehensive Clinical Nephrology, 7th Edition, Table 16.4
3. Rapidly Progressive GN (RPGN)
- Rapid loss of kidney function over days to weeks in the setting of nephritic syndrome - may present as a uremic emergency
- The histopathologic equivalent is crescentic glomerulonephritis (proliferative cells in Bowman's space forming a crescent shape)
- Can co-present with pulmonary hemorrhage as pulmonary-renal syndrome (ANCA-associated GN, anti-GBM/Goodpasture syndrome)
- Harrison's Principles of Internal Medicine 22E, p. 2458
Etiology-Based Classification
The preferred classification of GN is etiology-based because kidney pathology findings can overlap across causes. The major categories are:
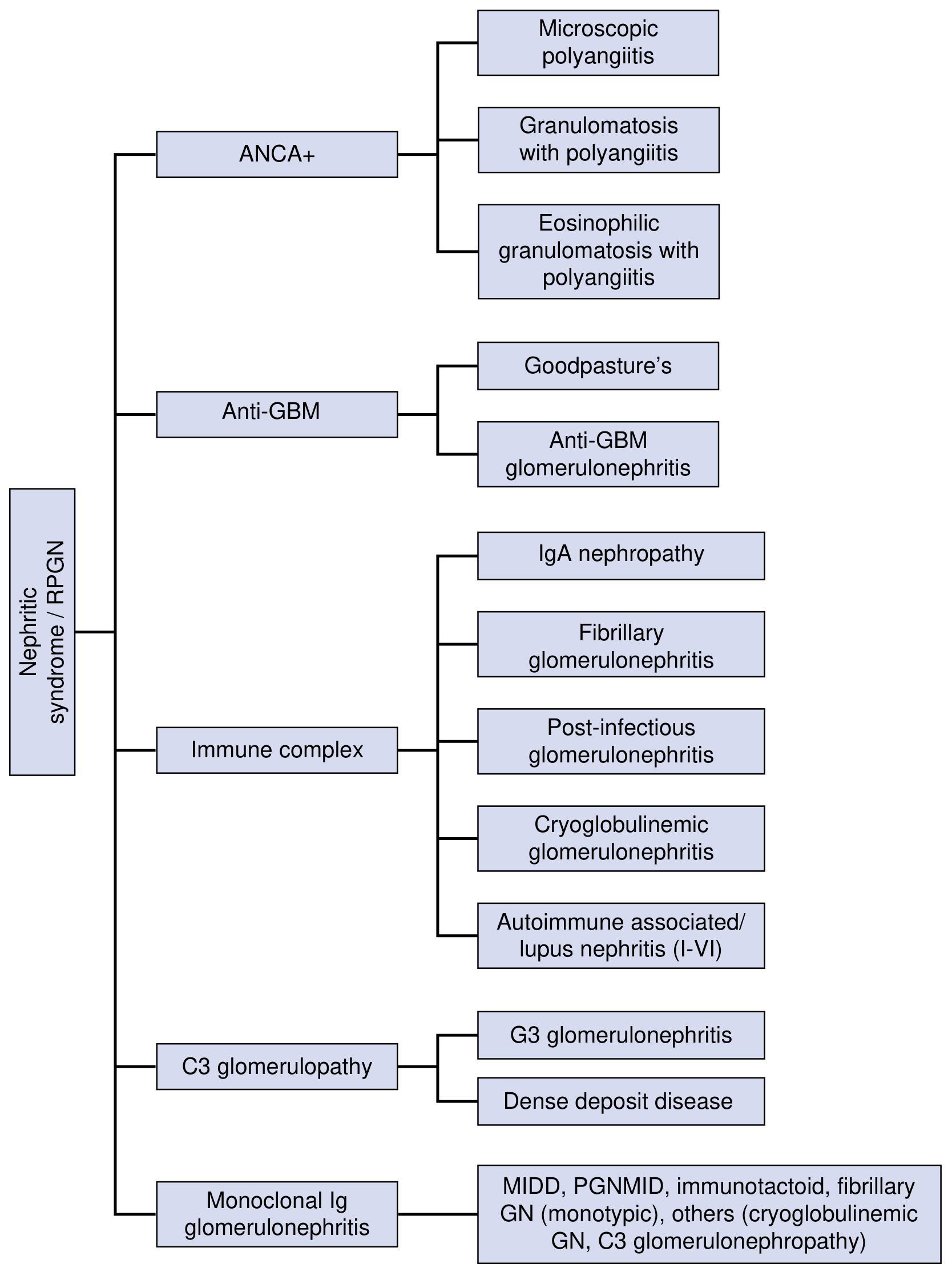
Fig. 16.3 - National Kidneys Foundation Primer on Kidney Diseases, 8e
A. Immune Complex-Mediated GN
Granular immune deposits (IgG, IgA, IgM, C3) in mesangium, subendothelial, or subepithelial locations.
1. Post-Infectious GN (PIGN)
- Occurs 1-4 weeks after streptococcal pharyngitis or impetigo (Group A beta-hemolytic strep, types 1, 4, 12)
- Most common in children aged 6-10 years
- Mechanism: deposition of immune complexes containing streptococcal pyogenic exotoxin B (SpeB) activates complement
- Low serum C3, elevated ASO/anti-DNase B titers
- Light microscopy: diffuse endocapillary proliferative GN with neutrophils in capillaries
- Immunofluorescence: granular IgG + C3
- Electron microscopy: subepithelial "humps" (pathognomonic)
- Treatment: supportive (treat underlying infection); generally self-limiting in children; worse prognosis in adults
2. IgA Nephropathy (Berger's Disease)
- Most common primary glomerulopathy worldwide
- Pathogenesis: deposition of galactose-deficient IgA1 immune complexes in the mesangium, triggered by upper respiratory infections (synpharyngitic hematuria)
- Presents with recurrent macroscopic or microscopic hematuria ± proteinuria
- Systemic form: IgA vasculitis (Henoch-Schönlein Purpura) - skin, joint, intestinal manifestations
- Light microscopy: MEST-C score (Mesangial proliferation, Endocapillary proliferation, Segmental sclerosis, Tubular atrophy/interstitial fibrosis, Crescents)
- Immunofluorescence: mesangial IgA deposits (hallmark)
- Prognosis: 60% benign; ~40% progress to ESKD over 10-20 years
- Predictors of progression: renal insufficiency, hypertension, proteinuria >1 g/24 hr at diagnosis; T score most reliable for ESKD
3. Lupus Nephritis
- ISN/RPS classification: Classes I-VI based on biopsy
- Classes III/IV (focal/diffuse proliferative) carry worst prognosis; Class V = membranous
- Low C3, C4; positive anti-dsDNA, ANA
4. Membranoproliferative GN (MPGN)
- "Tram-track" appearance on light microscopy (GBM duplication)
- Can present as nephrotic, nephritic, or mixed syndrome
5. Cryoglobulinemic GN - often HCV-associated
B. ANCA-Associated (Pauci-Immune) GN
- Absence of significant immune deposits on immunofluorescence
- Three entities:
- Microscopic polyangiitis (pANCA/MPO-ANCA)
- Granulomatosis with Polyangiitis (Wegener's) (cANCA/PR3-ANCA)
- Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss)
- Histology: necrotizing crescentic GN with few/no immune deposits
- Frequently presents as RPGN; can cause pulmonary-renal syndrome
C. Anti-GBM Disease (Type II RPGN)
- Autoantibodies against alpha-3 chain of type IV collagen (NC1 domain of GBM)
- Goodpasture disease = anti-GBM antibodies + RPGN + pulmonary hemorrhage
- Linear IgG deposits on immunofluorescence (distinctive pattern)
- Treatment: plasma exchange + high-dose steroids + cyclophosphamide
D. C3 Glomerulopathy
- Dysregulation of the alternative complement pathway
- Two subtypes: C3 glomerulonephritis and Dense Deposit Disease (DDD)
- Immunofluorescence: dominant C3 staining without significant immunoglobulin
E. Monoclonal Immunoglobulin-Associated GN
- Includes MIDD (monoclonal Ig deposition disease), PGNMID, immunotactoid GN, fibrillary GN (monotypic)
Diagnostic Work-Up
Urinalysis + microscopy:
- Dysmorphic RBCs, RBC casts, granular casts - hallmarks of glomerular disease
- Proteinuria quantification (spot urine protein:creatinine or 24-hour urine)
Serology panel (tailor to clinical context):
- ASO, anti-DNase B (post-infectious)
- ANA, anti-dsDNA, C3, C4 (lupus)
- ANCA (MPO, PR3)
- Anti-GBM antibodies
- Cryoglobulins, hepatitis B/C serology
- Serum protein electrophoresis/free light chains (monoclonal)
- Complement (C3/C4 low in immune complex GN, normal in pauci-immune)
Kidney biopsy - definitive diagnosis; assesses light microscopy, immunofluorescence (IF), and electron microscopy (EM)
Management Principles
General (disease-nonspecific):
- Low-salt, protein-restricted diet (0.8-1 g/kg/day)
- ACE inhibitor or ARB for anti-proteinuric effect and blood pressure control
- Diuretics for edema
- Statins for hyperlipidemia (nephrotic syndrome)
- Anticoagulation for high thrombotic risk (nephrotic, especially membranous/amyloidosis)
- SGLT-2 inhibitors - now have strong evidence for diabetic kidney disease and proteinuric GN
- National Kidneys Foundation Primer on Kidney Diseases, 8e
Disease-Specific:
| Cause | Treatment |
|---|---|
| Post-infectious GN | Supportive; antibiotics for active infection |
| IgA nephropathy | ACEi/ARB + immunosuppression for progressive disease |
| ANCA-associated GN | High-dose steroids + cyclophosphamide or rituximab |
| Anti-GBM disease | Plasma exchange + steroids + cyclophosphamide |
| Lupus nephritis (III/IV) | Mycophenolate or cyclophosphamide + steroids |
| MCD | Steroids (>90% response in children) |
| Membranous nephropathy | ACEi/ARB; rituximab for high-risk/refractory |
Key Distinguishing Features (Quick Reference)
| GN Type | IF Pattern | EM Finding | Serology |
|---|---|---|---|
| Post-streptococcal | Granular IgG + C3 | Subepithelial humps | Low C3, elevated ASO |
| IgA nephropathy | Mesangial IgA | Mesangial deposits | Normal complement |
| Anti-GBM | Linear IgG | GBM disruption | Anti-GBM Ab |
| ANCA-associated | Pauci-immune (negative) | No deposits | ANCA+ |
| Lupus nephritis | "Full house" (IgG, IgA, IgM, C3, C1q) | Subendothelial deposits | Low C3/C4, ANA+ |
| C3 GN/DDD | Dominant C3, no Ig | Dense intramembranous deposits (DDD) | Low C3, normal C4 |
Recent Evidence (2024-2026)
Recent systematic reviews provide updated insights:
- A 2026 meta-analysis on IgA nephropathy (PMID: 41385286) found that even low-grade proteinuria is associated with adverse kidney outcomes, reinforcing aggressive proteinuria management.
- A 2025 systematic review on IgA nephropathy prevalence in China (PMID: 40138167) highlights growing global burden.
- A 2024 meta-analysis on lupus nephritis glucocorticoid dosing (PMID: 38766897) shows that lower glucocorticoid doses achieve comparable complete response rates with fewer serious infections.
Sources: Comprehensive Clinical Nephrology 7e | National Kidneys Foundation Primer on Kidney Diseases 8e | Harrison's Principles of Internal Medicine 22E | Robbins & Cotran Pathologic Basis of Disease | Brenner and Rector's The Kidney
This is a shared conversation. Sign in to Orris to start your own chat.