Define copd exact from harrison
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
I have the exact definition from Harrison's. Here it is:
COPD - Definition from Harrison's Principles of Internal Medicine, 22nd Edition (2025)
Chapter 303 - Chronic Obstructive Pulmonary Disease
Craig P. Hersh, Edwin K. Silverman, Dawn DeMeo
"Chronic obstructive pulmonary disease (COPD) is defined as a disease state characterized by persistent respiratory symptoms and airflow obstruction.COPD includes:
- Emphysema - an anatomically defined condition characterized by destruction of the lung alveoli with air space enlargement
- Chronic bronchitis - a clinically defined condition with chronic cough and phlegm
- Small airway disease - a condition in which small bronchioles are narrowed and reduced in number
The classic definition of COPD requires the presence of chronic airflow obstruction, determined by spirometry, that usually occurs in the setting of noxious environmental exposures, most commonly products of combustion, including cigarette smoking in the United States and biomass fuels in some other countries.The increasing prevalence of vaping and use of inhaled cannabis are of increasing concern, especially in adolescent populations. Other factors including abnormal lung development, respiratory infections, asthma, and genetics, can lead to COPD.Emphysema, chronic bronchitis, and small airway disease are present in varying degrees in different COPD patients. Patients with a history of cigarette smoking without chronic airflow obstruction may have chronic bronchitis, emphysema, respiratory symptoms including dyspnea, and acute exacerbations. Although these patients are not included within the classic definition of COPD, they may have similar disease processes.Investigators in the COPDGene study proposed a multidimensional approach to COPD diagnosis, which is based on domains of: environmental exposures, respiratory symptoms, imaging abnormalities, and physiologic abnormalities."
Epidemiology (also from the same chapter):
- COPD is the 4th leading cause of death in the United States
- Affects >15 million persons in the United States
- Globally: an estimated 480 million individuals with COPD, projected to reach 592 million by 2050
- Harrison's Principles of Internal Medicine, 22nd Edition (2025), Chapter 303
Explain the pathogenesis and pathology of COPD
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
I now have all the content needed. Here is the full explanation directly from Harrison's 22nd Edition, Chapter 303.
Pathogenesis and Pathology of COPD
Harrison's Principles of Internal Medicine, 22nd Edition (2025) - Chapter 303
Overview
Airflow obstruction - the primary physiologic marker of COPD - results from airway disease and/or emphysema. Cigarette smoke exposure affects large airways, small airways (≤2 mm), and alveoli. Changes in large airways cause cough and sputum; changes in small airways and alveoli drive physiologic deterioration.
- Early COPD is driven by small airway disease
- Advanced COPD is characterized by extensive emphysema
- Greatest risk of progression: patients with both aggressive airway disease AND emphysema
1. Large Airway Disease (Chronic Bronchitis)
- Cigarette smoking causes mucus gland enlargement and goblet cell hyperplasia - producing the cough and phlegm that define chronic bronchitis
- Goblet cells increase in number and extend further down the bronchial tree
- Bronchi undergo squamous metaplasia, disrupting mucociliary clearance and predisposing to carcinogenesis
- Bronchial hyperreactivity (less than in asthma) can also cause airflow obstruction
- Mucus plugs (visible on CT) are associated with increased mortality
- Neutrophil elastase is a potent secretagogue, independent of its proteolytic activity
2. Small Airway Disease
The major site of increased airflow resistance is in airways ≤2 mm diameter. Key changes include:
- Goblet cell metaplasia - mucus-secreting cells replace surfactant-secreting club cells
- Smooth-muscle hypertrophy
- Luminal narrowing from fibrosis, excess mucus, edema, and cellular infiltration
- Reduced surfactant increases surface tension, predisposing to airway collapse
- Respiratory bronchiolitis with mononuclear inflammatory cells causes proteolytic destruction of elastic fibers at alveolar entrances
- Advanced COPD shows significant loss of smaller airways and loss of the lung microvasculature
Key sequence: Small airway narrowing and drop-out precede emphysematous destruction
3. Lung Parenchymal Destruction (Emphysema)
Emphysema = destruction of gas-exchanging air spaces (respiratory bronchioles, alveolar ducts, alveoli).
Cellular changes:
- Large numbers of macrophages accumulate in respiratory bronchioles of essentially all smokers
- Neutrophils, CD8+ T lymphocytes, and B lymphocytes are increased in the alveolar space
- Alveolar walls become perforated, then obliterated, coalescing into large emphysematous air spaces
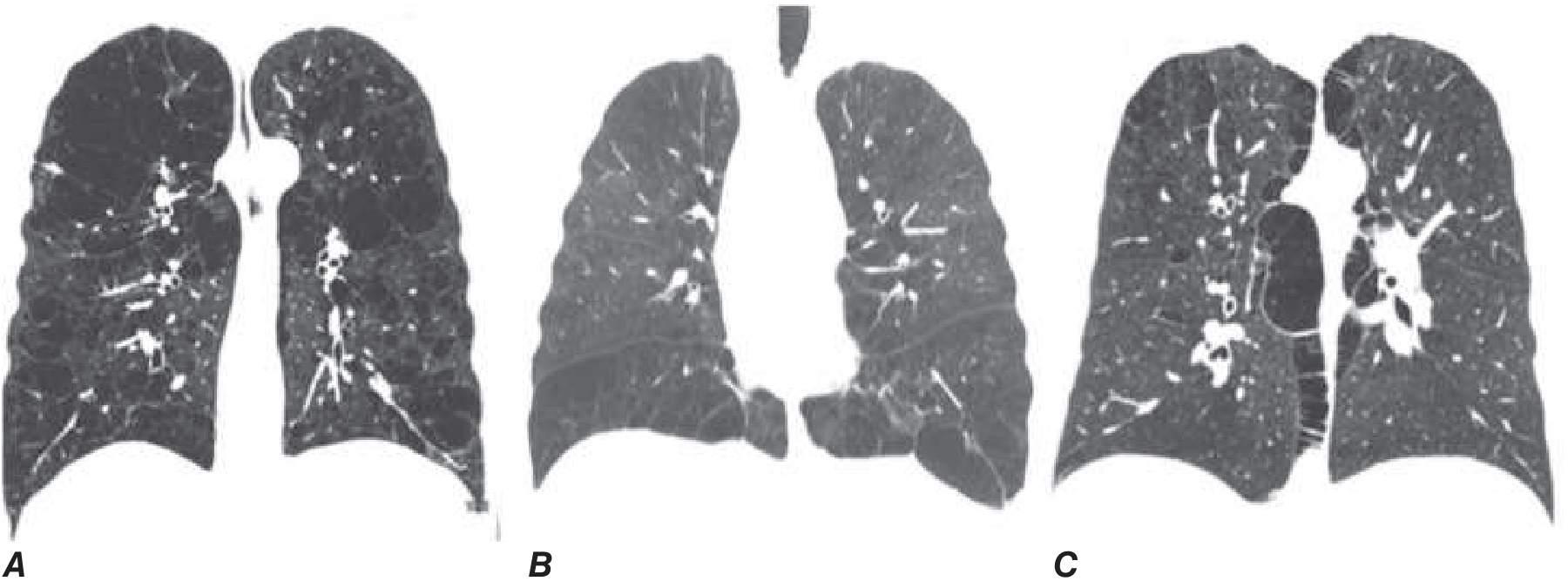
Pathologic types of emphysema (Figure 303-1):
| Type | Location | Association |
|---|---|---|
| Centrilobular | Around respiratory bronchioles; upper lobe predominant | Cigarette smoking (most common) |
| Panlobular | Evenly distributed across acinar units | Alpha-1 antitrypsin deficiency; lower lobe predominant |
| Paraseptal | Along pleural margins, sparing lung core | Peripheral distribution |
4. Disease Mechanisms - The Four-Step Paradigm
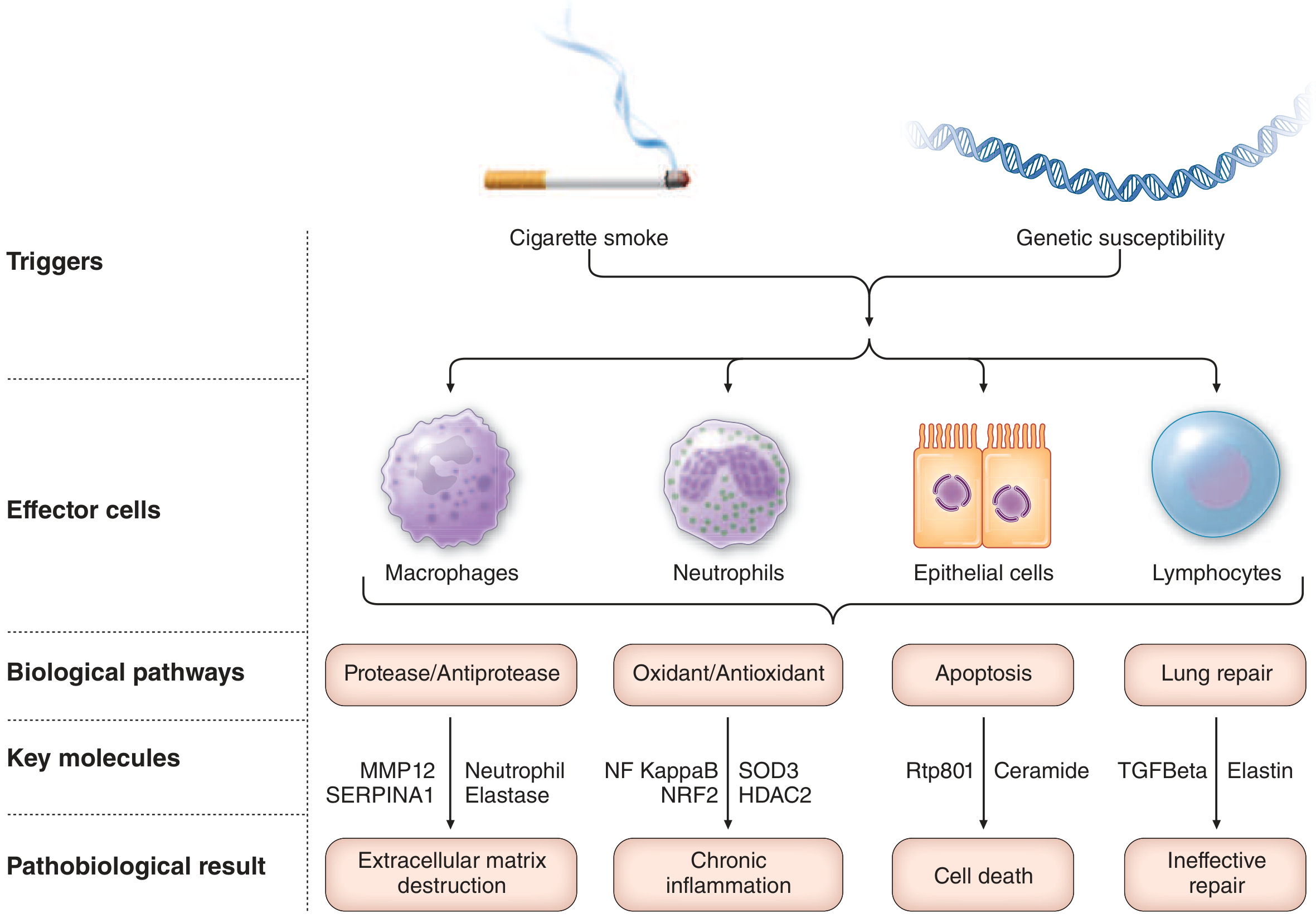
The dominant paradigm for COPD pathogenesis comprises four interrelated events:
- Inflammatory/immune cell recruitment - Chronic inhaled exposures in genetically susceptible individuals trigger recruitment of macrophages, neutrophils, CD8+ T lymphocytes, and epithelial cells to large airways, small airways, and terminal air spaces
- Proteinase release and extracellular matrix damage - Inflammatory cells release proteinases that damage the extracellular matrix supporting airways, vasculature, and gas-exchange surfaces
- Structural cell death - Via oxidant-induced damage, cellular senescence, and proteolytic loss of cellular-matrix attachments, leading to loss of smaller airways, vascular pruning, and alveolar destruction
- Disordered repair - Disordered repair of elastin and other extracellular matrix components contributes to air space enlargement and emphysema
Key biological pathways and molecules:
| Pathway | Key Molecules | Result |
|---|---|---|
| Protease/Antiprotease imbalance | MMP12, SERPINA1, Neutrophil elastase | Extracellular matrix destruction |
| Oxidant/Antioxidant imbalance | NF-kappaB, NRF2, SOD3, HDAC2 | Chronic inflammation |
| Apoptosis | Rtp801, Ceramide | Cell death |
| Lung repair | TGF-beta, Elastin | Ineffective repair |
Protease-Antiprotease Imbalance
- Elastin is a highly stable extracellular matrix component critical for lung integrity
- Proteolytic enzymes (especially MMP-12 from macrophages and neutrophil elastase) degrade elastin
- Alpha-1 antitrypsin (alpha1AT) normally protects against elastin destruction; its deficiency causes panlobular emphysema
Oxidant/Antioxidant Imbalance
- Cigarette smoke contains oxidants directly and stimulates macrophages/neutrophils to produce additional reactive oxygen species
- Oxidative stress activates NF-kappaB, driving chronic inflammation
- Reduced HDAC2 activity limits the ability of corticosteroids to suppress inflammation (explaining partial steroid resistance in COPD)
5. Physiologic Consequences
- Hypoxemia arises from ventilation-perfusion (V/Q) mismatch - not shunting; responds well to modest supplemental O2
- Hypercarbia (elevated PaCO2) does not typically occur until FEV1 < 25% of predicted
- Pulmonary arterial hypertension and cor pulmonale occur with FEV1 < 25% predicted AND chronic hypoxemia (PaO2 < 55 mmHg)
- Dynamic hyperinflation (air trapping) occurs due to loss of elastic recoil and airflow limitation, increasing the work of breathing
- Harrison's Principles of Internal Medicine, 22nd Edition (2025), Chapter 303, pp. 2292-2296
This is a shared conversation. Sign in to Orris to start your own chat.