Ewing Sarcoma
Reading File
Finding Sources
Searching PubMed
"Ewing sarcoma"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Let me compile the full answer.
Ewing Sarcoma
Overview
Ewing sarcoma is a malignant tumor of bone (and occasionally soft tissue) composed of small round blue cells, defined molecularly by translocations involving the EWSR1 gene on chromosome 22. It is the second most common primary bone sarcoma in children and adolescents (after osteosarcoma), and the most common primary bone malignancy in children under 10 years of age. The incidence is less than 1 per million per year, with roughly 200 new cases diagnosed annually in the United States.
Epidemiology
- Age: Most cases occur between ages 5-25 years; 80% of patients are under 20
- Sex: Slight male predominance
- Race: Exceedingly rare in individuals of African descent; predominantly affects Whites of European origin
- No known predisposing factors
Molecular Pathogenesis
The hallmark of Ewing sarcoma is a chromosomal translocation generating a chimeric fusion protein:
| Translocation | Genes Involved | Frequency |
|---|---|---|
| t(11;22)(q24;q12) | EWSR1 - FLI1 | ~85-90% of cases |
| t(21;22)(q22;q12) | EWSR1 - ERG | Less common |
| t(7;22)(p22;q12) | EWSR1 - ETV1 | Rare |
The resulting EWS/FLI1 chimeric protein binds to chromatin and dysregulates transcription, driving uncontrolled growth and abnormal differentiation. The cell of origin is uncertain but most likely mesenchymal stem cells or primitive neuroectodermal cells.
Location
- Long bones: Most common - metaphyses with frequent extension into the diaphysis; the classic description is "diaphyseal" but metaphyseal origin is more frequent
- Flat bones: Pelvis, ribs, shoulder girdle
- Spine: Primarily the sacrum (up to 50% of spinal cases), with posterior elements of the mobile spine commonly affected
- Extraskeletal (extraosseous): ~20% of cases arise in soft tissue
Clinical Presentation
- Pain - nearly universal; onset is insidious, often mild and intermittent initially
- Average diagnostic delay: ~34 weeks from symptom onset (15 weeks patient delay + 19 weeks physician delay)
- Soft tissue swelling over the affected site, often warm and tender
- Systemic symptoms mimicking osteomyelitis or infection: fever, erythema, leukocytosis, elevated ESR, elevated CRP
- Lab findings may include anemia and elevated inflammatory markers - leading to frequent initial misdiagnosis as osteomyelitis
Radiology
Plain Radiograph:
- Destructive lytic lesion with permeative ("moth-eaten") margins
- Classic "onion-skin" periosteal reaction - layers of reactive bone from tumor advancing through periosteum
- Large soft-tissue mass common
MRI (study of choice for local staging):
- Isointense to hyperintense on T2; isointense on T1
- Intense gadolinium enhancement due to hypercellularity
- "Curtain sign" - invasion of the spinal canal
- Paraspinal soft-tissue component often larger than the intraosseous lesion
- Should image the entire bone (not just the lesion) as large portions or even the whole bone may be involved
CT: Greater sensitivity than plain films for permeative appearance; chest CT mandatory to evaluate for pulmonary metastases (lung = most common metastatic site)
Bone Scan: Evaluates for bone metastases (2nd most common site)
FDG-PET/CT: Now the standard for initial staging and detection of recurrence; the initial SUV of the primary tumor correlates with tumor aggressiveness
Here is the MRI appearance of Ewing sarcoma of the L2 vertebral body, showing replacement of normal fatty marrow signal and paraspinal extension:
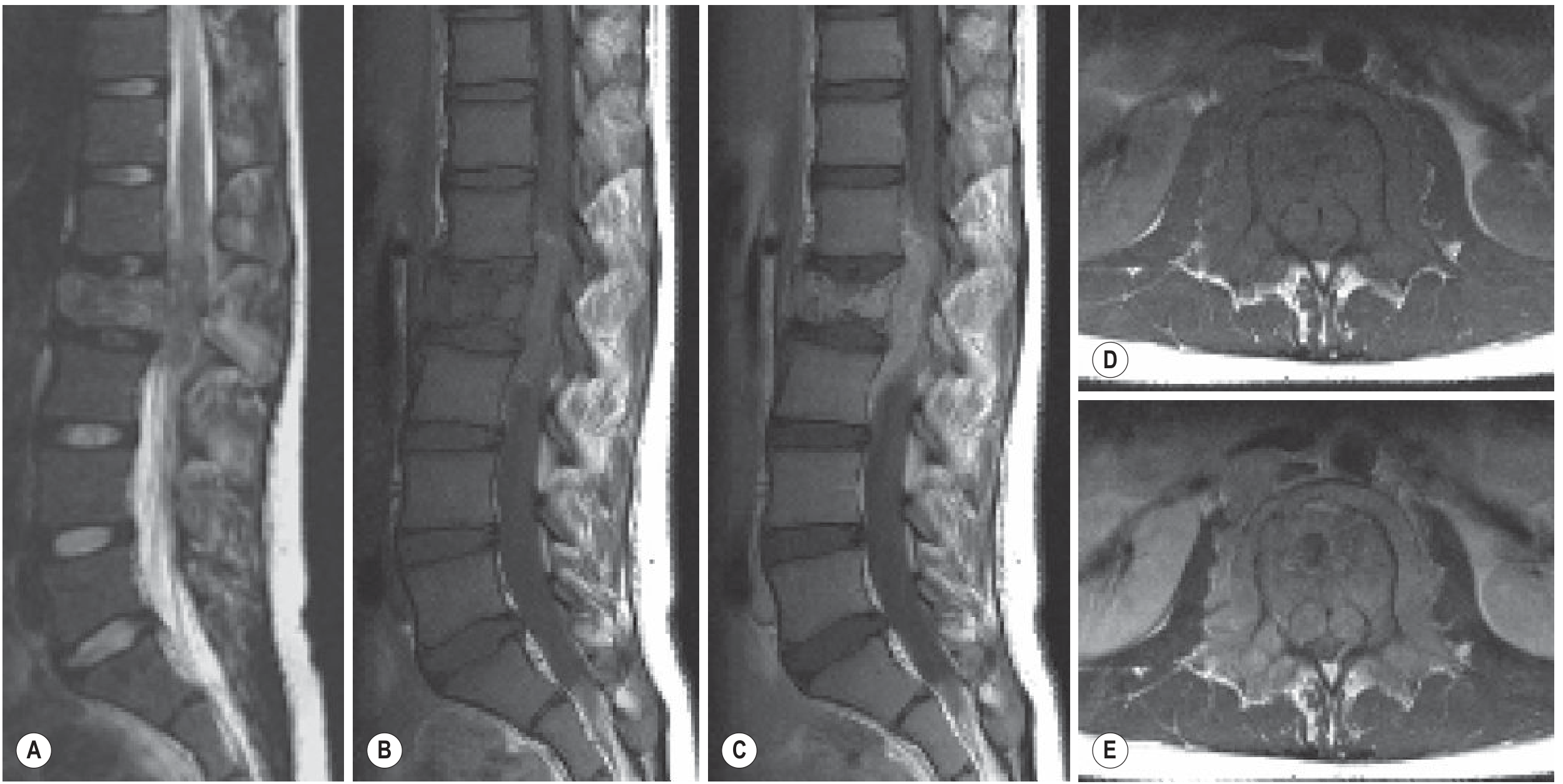
Pathology / Histology
Gross: Soft, tan-white tumor with areas of hemorrhage and necrosis. Arises in medullary cavity and invades cortex, periosteum, and soft tissue.
Microscopy:
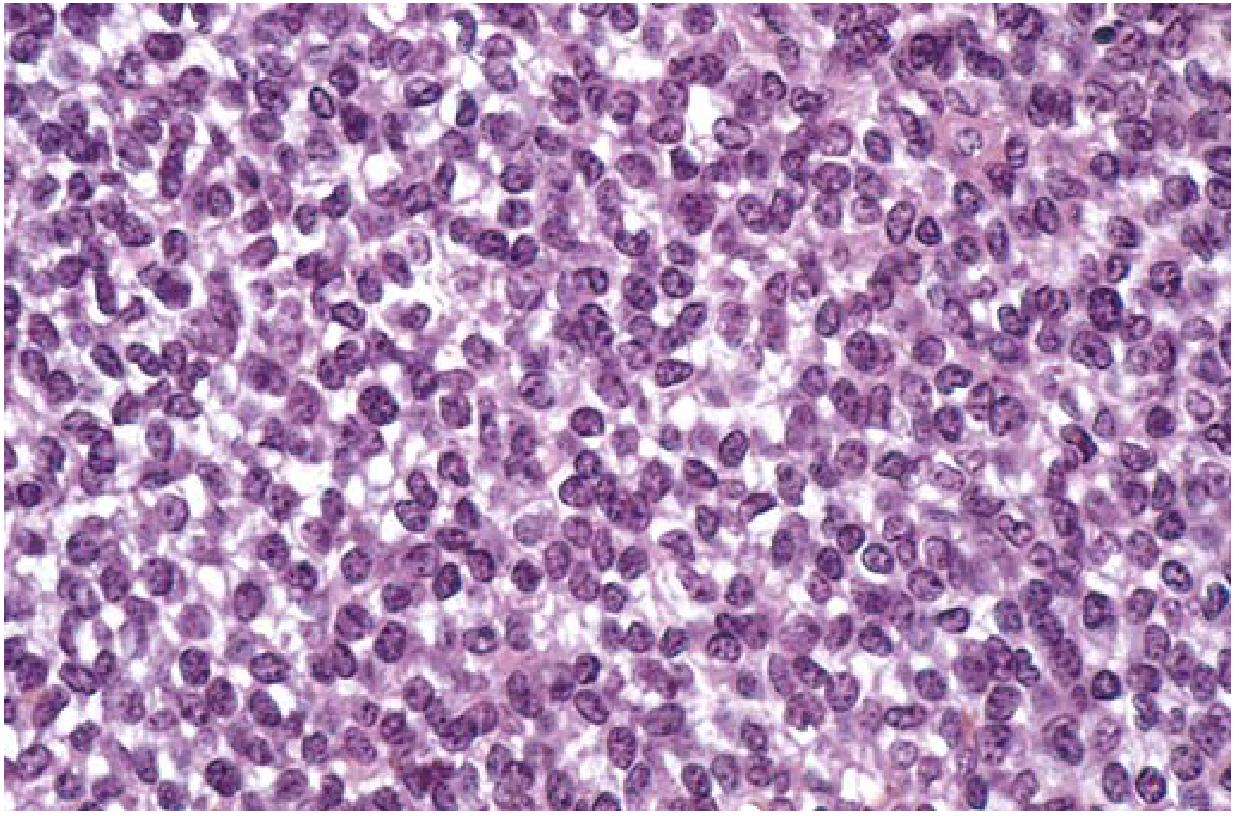
Fig. 19.23 - Ewing sarcoma composed of sheets of small round cells with minimal clear cytoplasm. (Robbins & Kumar Basic Pathology)
- Small round blue cells - sheets of uniform cells, slightly larger and more cohesive than lymphocytes
- Scant cytoplasm - appears clear due to intracellular glycogen
- Homer-Wright rosettes (circular groupings with central fibrillary core) - may be present
- Tumor cells do NOT produce bone or cartilage (distinguishes from osteosarcoma)
- PAS positive (glycogen), reticulin negative
- MIC-2 (CD99) positive by immunohistochemistry - relatively specific for Ewing sarcoma
Differential Diagnosis (Small Round Blue Cell Tumors of Childhood)
| Tumor | Distinguishing Feature |
|---|---|
| Ewing sarcoma | CD99+, PAS+, reticulin-, t(11;22) |
| Lymphoma | PAS-, reticulin+, LCA+, T/B cell markers+ |
| Embryonal rhabdomyosarcoma | Desmin+, myoglobin+, muscle-specific actins+ |
| Metastatic small cell carcinoma | Cytokeratin+ |
| Melanoma | Cytokeratin+, S100+ |
| Hemangiopericytoma | Factor VIII+ |
| Neuroblastoma | NSE+, catecholamines elevated |
Staging Workup
- Plain radiograph of primary lesion
- MRI of entire involved bone
- CT chest (lung metastases)
- Bone scan (skeletal metastases)
- FDG-PET/CT (preferred for full staging)
- Bone marrow aspirate (at some centers) to rule out diffuse systemic disease
Treatment
Treatment requires a multimodal approach. Before multiagent chemotherapy, long-term survival was <10%. Today, most centers report 60-75% long-term survival.
Chemotherapy (Neoadjuvant + Adjuvant)
The standard regimen is:
- VDC/IE alternating cycles:
- Vincristine + Doxorubicin + Cyclophosphamide (VDC)
- alternating with Ifosfamide + Etoposide (IE)
- Dose intensification has been shown to improve outcomes
- 5-year event-free survival with multiagent chemotherapy: ~70%
Local Control (Surgery vs. Radiation)
Ewing sarcoma is radiosensitive, but surgery is preferred when feasible:
- Wide surgical resection - associated with local recurrence rates <10% and improved overall survival; preferred for accessible lesions in expendable bones
- Radiation therapy - reserved for large, central, or unresectable tumors (e.g., pelvis, spine); also used when surgical margins are inadequate
- Decision is individualized; repeat staging after neoadjuvant chemotherapy guides surgical planning
- In growing children, wide resection avoids radiation-related complications (growth disturbance, secondary malignancies)
Metastatic / Relapsed Disease
- Prognosis is poor for patients with clinically detectable metastases at diagnosis
- Salvage regimens include:
- Cyclophosphamide + Topotecan
- Irinotecan + Temozolomide
- Gemcitabine + Docetaxel
Prognosis
| Disease Status | Outcome |
|---|---|
| Localized disease (neoadjuvant chemo + surgery ± radiation) | 75% 5-year survival; ~50% long-term cure |
| All patients with multiagent chemotherapy | ~70% 5-year event-free survival |
| Clinically detectable metastatic disease | Poor |
Favorable prognostic factors: Small tumor volume, distal extremity location, localized disease, good histologic response to neoadjuvant chemotherapy (>90% necrosis)
Unfavorable factors: Pelvic/axial location, large tumor volume, metastatic disease at presentation, older age, poor response to chemotherapy
Key High-Yield Points
- Translocation: t(11;22) - EWSR1-FLI1 fusion (>90% of cases)
- Histology: Small round blue cell tumor; PAS+, CD99+, reticulin-
- Radiograph: Lytic, permeative lesion + "onion-skin" periosteal reaction
- Most common site of metastasis: Lungs (then bone)
- Treatment backbone: VDC/IE chemotherapy + surgery ± radiation
- Mimics osteomyelitis: Fever, leukocytosis, elevated ESR/CRP
- Rare in African descendants; predominantly European-descent population
Sources: Robbins & Kumar Basic Pathology, p. 786 | Goldman-Cecil Medicine, p. 2115 | Campbell's Operative Orthopaedics 15th Ed 2026, p. 1221-1222 | Grainger & Allison's Diagnostic Radiology, p. 1285 | Miller's Review of Orthopaedics 9th Ed
This is a shared conversation. Sign in to Orris to start your own chat.