Difference between nephrogenic and central Diabetes insipidus
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have enough comprehensive information to give a thorough answer. Let me compile it:
Central vs. Nephrogenic Diabetes Insipidus
Both forms share the same hallmark presentation - polyuria (>50 mL/kg/day of hypotonic urine), polydipsia, and dilute urine - but the underlying defect, causes, diagnostic response, and treatment differ fundamentally.
Core Pathophysiology
| Feature | Central DI | Nephrogenic DI |
|---|---|---|
| Defect location | Hypothalamus / posterior pituitary | Kidney (collecting duct) |
| Mechanism | Failure to synthesize or secrete ADH (vasopressin) | Renal resistance/unresponsiveness to ADH |
| ADH levels | Low or absent (unmeasurable) | Normal or elevated |
| Aquaporin-2 (AQP2) | AQP2 present but not stimulated | AQP2 absent, mutated, or signaling is blocked |
In central DI, as few as 10-15% of normal vasopressinergic neurons in the hypothalamus are sufficient to maintain asymptomatic urine volumes - loss of just a few more neurons produces rapid symptomatic polyuria. In nephrogenic DI, vasopressin is secreted normally from the posterior pituitary, but the principal cells of the collecting duct cannot respond to it. - Goldman-Cecil Medicine, p. 2190-2194
Causes
Central DI (acquired > congenital)
- Tumors: craniopharyngioma, germinoma, pinealoma, metastases (lung, breast)
- Trauma: head injury, basal skull fracture, post-neurosurgical (especially after pituitary adenoma resection)
- Infiltrative/inflammatory: neurosarcoidosis, histiocytosis (Langerhans cell), tuberculous meningitis, multiple sclerosis
- Vascular: hypothalamic hemorrhage
- Idiopathic: up to 30-50% of cases (not considered truly idiopathic until 4 years of follow-up with negative annual imaging)
- Genetic (autosomal dominant): mutations in the signal peptide or neurophysin portion of the pre-prohormone; onset is typically delayed to childhood - Goldman-Cecil Medicine, p. 2192-2194
Nephrogenic DI (renal resistance)
- Genetic (congenital):
- X-linked (>90% of congenital cases): mutations in the AVPR2 gene (vasopressin V2 receptor); affects males, frequency 4-8 per million male births; presents in the first week of life with vomiting, failure to thrive, hypernatremia
- Autosomal recessive/dominant (<10%): mutations in the AQP2 gene (aquaporin-2 water channel)
- Acquired (more common than congenital):
- Drugs: lithium (most common - blocks adenylyl cyclase in collecting duct cells), demeclocycline, amphotericin B
- Electrolyte disorders: hypokalemia, hypercalcemia
- Renal diseases: sickle cell disease, sarcoidosis, pyelonephritis, multiple myeloma, analgesic nephropathy, ureteric obstruction, CKD
- Diuretics (furosemide): impair loop of Henle concentrating mechanism - Guyton & Hall Physiology, p. 380; Goldman-Cecil Medicine, p. 2321
Clinical Features
| Feature | Central DI | Nephrogenic DI |
|---|---|---|
| Urine volume | Up to 18-24 L/day (complete) | Large, but may be less extreme in partial/acquired forms |
| Onset | Often relatively abrupt (days to weeks) | Congenital: neonatal period; Acquired: gradual |
| Thirst | Present and strong (preference for cold liquids) | Present; thirst mechanism intact |
| Nocturia/noctural polydipsia | Yes - disrupts sleep | Yes |
| Serum sodium | High-normal (mild hypernatremia if thirst insufficient) | Can be significantly elevated in congenital neonatal form |
| Uric acid | Elevated (>5 mg/dL) - helps distinguish from primary polydipsia | Similar |
| Bright spot on T1 MRI | Absent (normally present in posterior pituitary) | Present (normal posterior pituitary) |
Diagnostic Workup
The diagnostic flowchart below (from Goldman-Cecil Medicine) outlines the approach to polyuria:
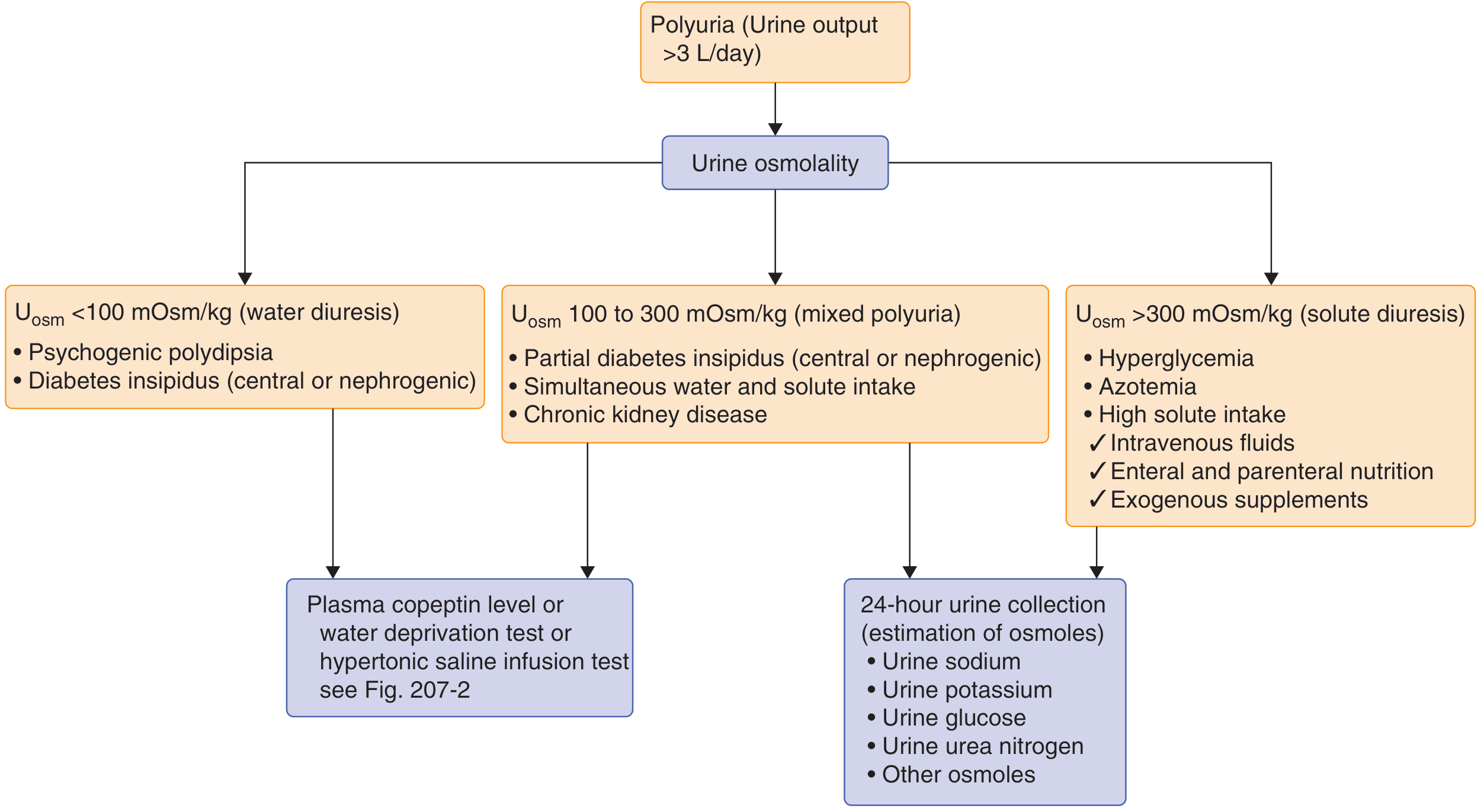
Both types produce urine osmolality <100 mOsm/kg (water diuresis). The key distinguishing test is the desmopressin (DDAVP) test:
Water Deprivation + Desmopressin Test
| Response after Desmopressin | Diagnosis |
|---|---|
| Urine osmolality increases >50% | Complete central DI |
| Urine osmolality increases 10-50% | Partial central DI |
| No significant increase (<10%) in urine osmolality | Nephrogenic DI |
| No increase (already maximally concentrated during deprivation) | Primary polydipsia |
Lack of a prompt decrease in urine volume and increase in urine osmolarity within 2 hours after desmopressin injection is strongly suggestive of nephrogenic DI. - Guyton & Hall Physiology, p. 380
Plasma Copeptin
A newer test (replacing water deprivation in many centers): copeptin (C-terminal fragment of the vasopressin precursor) is low in central DI and high or normal in nephrogenic DI and primary polydipsia.
Treatment
Central DI
- Desmopressin (DDAVP) - synthetic ADH analogue; drug of choice
- Intranasal: 5-20 µg per dose (spray or rhinal catheter)
- Oral: start with 0.05 mg (half a 0.1 mg tablet) at bedtime; titrate to 8-12 hour dosing
- Parenteral (hospitalized patients): 1-2 µg IV/SC every 8-12 hours
- Monitor serum sodium to avoid hyponatremia from over-treatment
- Treat the underlying cause (surgery, radiation for tumors)
- Post-surgical DI often recovers within 1 year; weekly dose holds can assess ongoing need - Goldman-Cecil Medicine, p. 2337-2341
Nephrogenic DI
- Address underlying cause: stop offending drugs (lithium, demeclocycline), correct hypokalemia/hypercalcemia
- Thiazide diuretics (paradoxically reduce polyuria): cause mild sodium depletion, reduce GFR, increase proximal tubule reabsorption - less fluid reaches the collecting duct
- Low-sodium, low-protein diet: reduces osmotic load
- NSAIDs (indomethacin): inhibit prostaglandins that antagonize ADH action - used adjunctively
- Amiloride: used specifically for lithium-induced nephrogenic DI - blocks lithium entry into collecting duct cells
- Desmopressin is ineffective in complete nephrogenic DI (the receptor/signaling pathway is the problem)
Summary Table
| Parameter | Central DI | Nephrogenic DI |
|---|---|---|
| Site of defect | Hypothalamus/pituitary | Kidney (collecting duct) |
| ADH secretion | Absent or low | Normal or high |
| ADH action | Cannot be tested (none released) | Defective despite normal/high ADH |
| Serum ADH/copeptin | Low | Normal or elevated |
| Response to desmopressin | Responds (urine concentrates) | Does not respond |
| MRI posterior pituitary | Bright spot absent | Bright spot present |
| Genetics | Autosomal dominant (AVPR gene mutations) | X-linked (AVPR2) or AR/AD (AQP2) |
| Treatment | Desmopressin | Thiazides, NSAIDs, amiloride, low-Na diet |
Sources: Goldman-Cecil Medicine (International Edition), Guyton & Hall Textbook of Medical Physiology, Tietz Textbook of Laboratory Medicine 7th Ed, Costanzo Physiology 7th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.