lupus
Reading File
Finding Sources
Searching PubMed
"systemic lupus erythematosus"[MeSH Terms] AND treatment
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Here is a comprehensive overview of Systemic Lupus Erythematosus (SLE).
Systemic Lupus Erythematosus (SLE)
What Is It?
SLE is a chronic, multisystem autoimmune disease of unknown cause characterized by the production of autoantibodies against numerous self-antigens, particularly nuclear components. The immune complexes that form deposit in tissues and trigger widespread inflammation and vasculitis, leading to multiorgan damage. The disease follows an unpredictable course with flares and remissions.
Epidemiology
- Affects approximately 1 in 1,000-2,500 people in the general population
- Far more common in African American women (up to 1 in 250, ages 18-65)
- Predominantly affects women of reproductive age (15-40 years)
- Female:male ratio is 2:1 before puberty and 4:1 after puberty
- Strong familial tendency; associated with MHC genes DR2, DR3, DR4, and DR5
- Environmental triggers include infections, UV sunlight, stress, drugs, and toxins
Pathophysiology
The central defect is loss of self-tolerance, driven by:
- B cell overactivity producing antinuclear antibodies (ANAs)
- The most disease-specific antibodies are anti-dsDNA and anti-Sm (Smith antigen) - found almost exclusively in SLE
- Immune complex deposition activates complement and causes end-organ inflammation
- T cell, B cell, and monocyte dysfunction all contribute
Clinical Features
SLE is truly systemic. Constitutional symptoms (fatigue, malaise, fever, weight loss) are common. The main organ systems affected:
Mucocutaneous (>90% of patients)
- Malar (butterfly) rash - fixed erythema over cheeks sparing the nasolabial folds; present in ~1/3 of patients; often triggered by sun exposure
- Subacute cutaneous LE (SCLE) - annular or papulosquamous lesions on sun-exposed areas; associated with anti-Ro/SS-A antibodies in ~70%; does NOT scar
- Discoid LE (DLE) - raised erythematous plaques with thick scales; typically on face, neck, scalp, ears; DOES cause scarring with hypopigmented atrophic centers
- Photosensitivity (1/3 to 2/3 of patients)
- Oral/nasal/vaginal ulcers, alopecia, palpable purpura
Joints
- Nonerosive arthritis of 2+ peripheral joints (tenderness, swelling, effusion)
Kidneys (Lupus Nephritis)
- Persistent proteinuria >0.5 g/day, or cellular casts
- One of the most serious complications
Cardiovascular / Serositis
- Pleuritis, pericarditis
- Antiphospholipid antibody syndrome: venous/arterial thrombosis, livedo reticularis
Neurologic
- Seizures or psychosis (in the absence of metabolic causes)
Hematologic
- Hemolytic anemia, leukopenia (<4000/mm³), lymphopenia (<1500/mm³), thrombocytopenia (<100,000/mm³)
Diagnostic Criteria (ACR - 11 Criteria)
≥4 of 11 criteria required (present serially or simultaneously):
| # | Criterion | Details |
|---|---|---|
| 1 | Malar rash | Fixed erythema over malar eminences, spares nasolabial folds |
| 2 | Discoid rash | Erythematous raised patches, scaling, follicular plugging, scarring |
| 3 | Photosensitivity | Rash from sunlight exposure |
| 4 | Oral ulcers | Nasal or oral ulceration, usually painless |
| 5 | Arthritis | Nonerosive, 2+ peripheral joints |
| 6 | Serositis | Pleuritis or pericarditis |
| 7 | Renal disorder | Proteinuria >0.5 g/day or cellular casts |
| 8 | Neurologic | Seizures or psychosis (not drug/metabolic-related) |
| 9 | Hematologic | Hemolytic anemia, leukopenia, lymphopenia, or thrombocytopenia |
| 10 | Immunologic | Anti-dsDNA, anti-Sm, or false-positive syphilis test |
| 11 | ANA | Abnormal titer in absence of drug-induced lupus |
Laboratory Findings
- ANA - positive in >95% of SLE; highly sensitive but not specific
- Anti-dsDNA and anti-Sm - highly specific for SLE
- Anti-Ro/SS-A and anti-La/SS-B - associated with SCLE and neonatal lupus
- Complement (C3, C4) - low during active disease
- Anemia, leukopenia, thrombocytopenia on CBC
- Elevated creatinine, proteinuria, hematuria in renal involvement
- False-positive VDRL/RPR (syphilis test)
Approach to Cutaneous Lupus
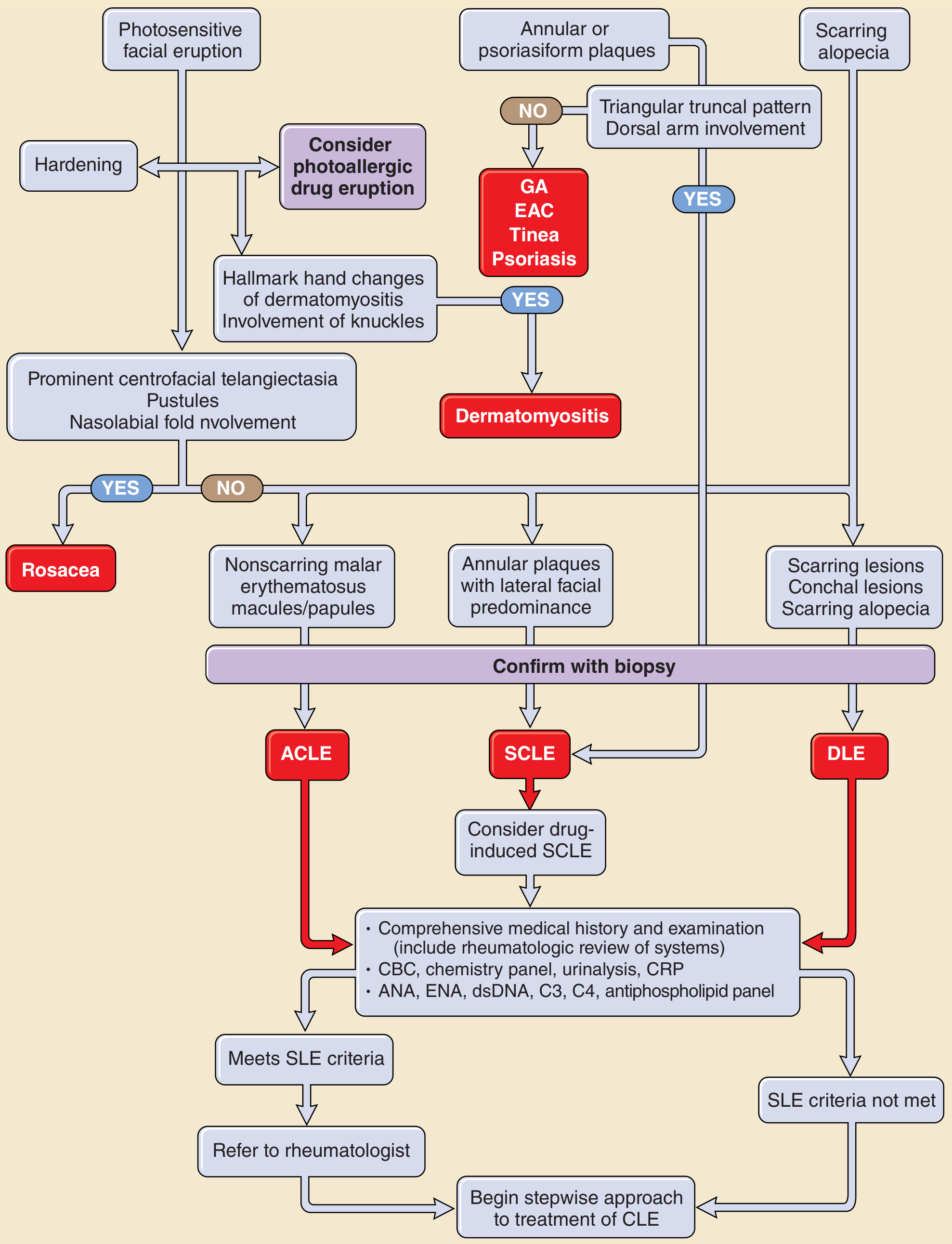
Figure: Approach to the patient with skin lesions suspicious for cutaneous lupus (ACLE = acute cutaneous LE, SCLE = subacute cutaneous LE, DLE = discoid LE) - Fitzpatrick's Dermatology
Management
General Principles
- Avoid precipitating factors, especially sun exposure (use sunscreen, protective clothing)
- Monitor for organ involvement at each visit
- Long-term physician-patient relationship between family doctor and rheumatologist is important
Pharmacologic Treatment by Severity
Mild disease (skin, joints, serositis):
- NSAIDs - first-line for myositis, serositis, arthritis
- Topical corticosteroids - for cutaneous lupus (SOR: B)
- Hydroxychloroquine (antimalarial) - for persistent cutaneous lesions; also reduces flares and has cardioprotective effects
Moderate-Severe disease:
- Prednisone 0.5 mg/kg/day for arthritis/serositis/constitutional symptoms
- Prednisone 1 mg/kg/day for nephritis and CNS disease
- IV methylprednisolone for severe hemolytic anemia
Refractory or steroid-resistant:
- Immunosuppressives: cyclophosphamide, azathioprine, mycophenolate mofetil (especially for lupus nephritis)
- If no steroid response after 7 weeks, add immunosuppressive therapy
Antiphospholipid syndrome:
- Warfarin (INR 2-3) for venous/arterial thrombosis; often lifelong
End-stage lupus nephropathy:
- Dialysis or kidney transplantation
Outcome
- 5-year survival: 90% with modern treatment
-
90% of treated patients survive at least 15 years
- SLE was once considered a universally progressive, terminal disease; outcomes have dramatically improved
Lupus Variants
| Type | Features |
|---|---|
| Discoid LE (DLE) | Chronic, scarring skin-only disease; <5% progress to SLE |
| Subacute cutaneous LE (SCLE) | Nonscarring, sun-exposed; anti-Ro positive; ~15% have SLE |
| LE Panniculitis / Profundus | Deep subcutaneous nodules; ~50% have SLE |
| Chilblain LE | Cold-triggered purple-red papules on fingers/toes; ~20% develop SLE |
| Drug-induced lupus | Resolves when drug stopped; anti-histone antibodies |
| Neonatal lupus | From maternal anti-Ro/La transfer; congenital heart block |
Sources: Textbook of Family Medicine 9e, p. 899-902 | Fitzpatrick's Dermatology, Vol. 1-2 | Andrews' Diseases of the Skin
sjogren syndrome
Finding Sources
Searching PubMed
"Sjogren syndrome"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Here is a comprehensive overview of Sjögren Syndrome drawn from multiple authoritative textbooks.
Sjögren Syndrome
Definition
Sjögren syndrome (SS) is a chronic autoimmune disorder of the exocrine glands, characterized by lymphoplasmacytic infiltration of the salivary and lacrimal glands, causing glandular hypofunction. The hallmark result is the sicca complex: dry mouth (xerostomia) and dry eyes (keratoconjunctivitis sicca).
Primary vs. Secondary
| Type | Definition |
|---|---|
| Primary SS | Sicca symptoms as the isolated disease |
| Secondary SS | Sicca symptoms occurring alongside another autoimmune disease - RA, SLE, polymyositis, systemic sclerosis, or primary biliary cirrhosis |
Epidemiology
- Estimated prevalence: 1-3% of the population
- Most common in the 4th to 5th decades of life
- >90% of patients are women
- Associated with HLA B8 and DR3
Pathophysiology
The trigger is likely an initial viral infection in a genetically susceptible host (HLA-B8/DR3), leading to:
- Aberrant autoimmune activation
- Dense lymphocytic infiltration of exocrine glands
- B-cell overstimulation - excess immunoglobulins and autoantibodies; autoreactive B-cell clones escape tolerance checkpoints
- Formation of germinal centers within salivary glands
- Increased follicular helper T cells driving B-cell responses
- Production of characteristic autoantibodies: anti-Ro/SS-A and anti-La/SS-B
Clinical Features
Sicca (Glandular) Manifestations
Xerostomia (dry mouth):
- Difficulty chewing, swallowing, and speaking
- Intolerance to acidic/spicy foods
- Dental caries (accelerated, due to reduced saliva)
- Adherence of food to buccal mucosa
- Smooth, fissured tongue with papillary atrophy
- Absence of pooled saliva in floor of mouth
- Scant or cloudy saliva from ducts
- Candida albicans overgrowth (common)
- Parotid gland enlargement in 25-66% of patients - may be unilateral initially, usually becomes bilateral; episodic or chronic
Keratoconjunctivitis sicca (dry eyes):
- Foreign body sensation - "gritty" or "sandy" feeling
- Dilated bulbar conjunctival vessels, periorneal injection
- Corneal/conjunctival epithelial destruction
- Lacrimal gland enlargement (occasionally)
Systemic (Extraglandular) Manifestations
Occur in approximately one-third of patients:
| System | Manifestations |
|---|---|
| Constitutional | Fatigue, low-grade fever, malaise, myalgia, arthralgia |
| Respiratory | Pharyngeal/esophageal dryness, dysphagia; tracheobronchial involvement - bronchitis, pneumonia |
| Renal | Tubulointerstitial nephritis, distal renal tubular acidosis, impaired concentrating ability, hypercalciuria; glomerulonephritis in ~4% (rare) |
| Vascular | Vasculitis in 20-30% - Raynaud phenomenon, recurrent urticaria-like lesions |
| Neurologic | Peripheral sensory and motor polyneuropathies; can mimic multiple sclerosis; CNS involvement reported |
| Lymphoproliferative | Increased lymphoma risk (even after decades of benign disease); persistent parotid enlargement is a warning sign |
Diagnosis
Objective Tests
| Test | What It Measures |
|---|---|
| Schirmer II test | Nasolacrimal reflex tear production; <8 mm wetting in 5 min = abnormal |
| Rose Bengal staining | Damaged corneal/conjunctival epithelium |
| Salivary flow rate | Measured with Lashley cups over Stensen duct opening |
| Sialography | Sialectasis in 85-97% of SS patients |
| Minor salivary gland biopsy | Required for definite diagnosis - shows focal mononuclear aggregates (chronic focal sialadenitis); focus score ≥1 per 4 lobes |
Serology
- Anti-Ro/SS-A - most sensitive (70-90% in SCLE overlap; present in SS)
- Anti-La/SS-B - more specific, less sensitive
- ANA - positive in 60-80%
- Elevated RF (>1:320) in many patients
- Hypergammaglobulinemia, cryoglobulins
- Complement (C3/C4) - generally normal in SS (unlike SLE, where it is low)
- B-cell subset analysis recommended
San Diego Criteria for Definite SS
All three of the following must be met:
- Objective signs of ocular dryness (Schirmer <8 mm AND positive Rose Bengal)
- Objective signs of dry mouth (reduced parotid flow AND positive minor salivary gland biopsy - focus score ≥1)
- Serologic evidence of systemic autoimmunity (elevated RF >1:320, ANA >1:320, or anti-SS-A/SS-B)
Probable SS = criteria met without salivary gland biopsy (decreased salivary function + ocular + serologic)
Exclusions
HIV, primary fibromyalgia, sarcoidosis, preexisting lymphoma, hepatitis B or C, other known causes of keratitis sicca or salivary gland enlargement
Differential Diagnosis
- Medications (most common cause of xerostomia) - sedatives, antipsychotics, antidepressants, antihistamines, diuretics
- Sarcoidosis, lymphoma
- HIV, hepatitis C infection
- Radiation sialadenitis (>4,000 cGy causes permanent secretory hypofunction)
- Diabetes mellitus, cystic fibrosis
- Bell palsy, MS (neuropathic glandular dysfunction)
Management
Ocular (Dry Eye)
- Artificial tears (monitor for blepharitis from preservatives)
- Punctal plugs - collagen (temporary, dissolve in 2 days) or silicone (durable, removable); can progress to permanent punctal occlusion surgery
- Cyclosporine ophthalmic drops - FDA approved for keratoconjunctivitis sicca; objective and subjective improvement
- Humidifiers
- Patching for corneal ulceration
Oral (Dry Mouth)
- Frequent sips of water; sugarless gum or mints (stimulate residual function)
- Artificial saliva (Salivart, Mouth Kote)
- Pilocarpine (muscarinic agonist) 5 mg three to four times daily - most used systemic sialagogue; side effects: flushing, sweating, urinary frequency
- Cevimeline (also a muscarinic agonist) - alternative secretagogue
- Meticulous fluoride dental care to prevent caries
- Topical antifungal troches for Candida overgrowth
Systemic Disease
- NSAIDs and hydroxychloroquine (antimalarials) - for arthralgias and systemic symptoms
- Corticosteroids - for tubulointerstitial nephritis (usually responds well), and severe extraglandular complications
- Immunosuppressives (cyclophosphamide, azathioprine) - for refractory severe disease, necrotizing vasculitis
- Rituximab (anti-CD20) - B-cell depletion therapy; improves xerostomia, fatigue, and joint tenderness; not beneficial for low disease activity alone; used with corticosteroids for systemic involvement
- Belimumab and abatacept - small preliminary trials show promising results
- Sialendoscopy - for recurrent sialadenitis; duct dilation and washout; may allow lower-intensity medical therapy
Lymphoma Surveillance
Patients with persistent unilateral or bilateral parotid enlargement are at higher risk for lymphoma development and require monitoring.
Sources: Textbook of Family Medicine 9e, p. 902-903 | Cummings Otolaryngology Head and Neck Surgery | Brenner and Rector's The Kidney
This is a shared conversation. Sign in to Orris to start your own chat.