
17819571069104339345939112547676.jpg
Parathyroid gland (more focus on hyperparathyroidism)
Reading File
Finding Sources
Searching PubMed
"hyperparathyroidism"[MeSH Terms] AND "primary hyperparathyroidism"[MeSH Terms]
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive content from Robbins & Cotran. Let me compile the full, detailed answer.
Parathyroid Glands and Hyperparathyroidism
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease, pp. 1010-1013
1. Normal Parathyroid Gland Anatomy & Histology
The parathyroid glands develop from the pharyngeal pouches (same pouches that give rise to the thymus). There are four glands, located near the upper and lower poles of each thyroid lobe, but ectopic locations are possible - the carotid sheath, thymus, and anterior mediastinum.
Two cell types:
| Cell Type | Characteristics | Function |
|---|---|---|
| Chief cells | Predominate; 12-20 µm, polygonal, round central nuclei, light-to-dark pink cytoplasm | Contain secretory granules with PTH |
| Oxyphil cells | Slightly larger, eosinophilic cytoplasm, packed with mitochondria, sparse secretory granules | Large number of calcium-sensing receptors; produce PTHrP and calcitriol |
In early childhood, glands are almost entirely chief cells. Stromal fat increases until age ~25, reaching a maximum of ~30% of gland volume.
2. PTH - Functions (Calcium Homeostasis)
The parathyroids respond to free (ionized) calcium in the blood. Low calcium → PTH release. PTH acts via four mechanisms:
- Renal tubular reabsorption of calcium (conserving free Ca²⁺)
- Activates vitamin D (1α-hydroxylase in kidney) → increases intestinal calcium absorption
- Increases urinary phosphate excretion → lowers serum phosphate → further raises ionized Ca²⁺
- Stimulates osteoclast differentiation → bone resorption → releases ionized calcium
The net effect is elevated free calcium, which then inhibits further PTH secretion via classic negative feedback.
3. Hyperparathyroidism - Overview
Hyperparathyroidism is classified into three types:
| Type | Mechanism |
|---|---|
| Primary | Autonomous overproduction of PTH (adenoma/hyperplasia) |
| Secondary | Compensatory hypersecretion due to chronic hypocalcemia (renal failure, Vit D deficiency) |
| Tertiary | Autonomous PTH production developing after long-standing secondary HPT |
4. Primary Hyperparathyroidism (1° HPT)
Epidemiology
- One of the most common endocrine disorders
- Annual incidence: ~25 cases per 100,000 in US/Europe
- Female:male ratio = nearly 4:1
- Most patients are in their 50s or older
- Up to 80% are asymptomatic - discovered incidentally on serum electrolyte panels
Causes (by frequency)
- Adenoma: 85-95% (solitary, almost always sporadic)
- Primary hyperplasia (diffuse or nodular): 5-10%
- Parathyroid carcinoma: ~1%
Molecular Pathogenesis of Sporadic Adenomas
1. Cyclin D1 (CCND1) gene inversions:
A pericentric inversion on chromosome 11 places CCND1 (normally on 11q) adjacent to the PTH gene (on 11p). The PTH gene's regulatory elements then drive overexpression of cyclin D1, a major cell cycle regulator - causing uncontrolled proliferation. Cyclin D1 is overexpressed in ~40% of parathyroid adenomas even without CCND1 rearrangement.
2. MEN1 mutations:
~15% of sporadic tumors have somatic mutations in the MEN1 tumor suppressor gene (chromosome 11q13) with loss of heterozygosity (LOH). Germline MEN1 mutations cause MEN-1 syndrome.
3. CDC73 mutations:
Mutated in ~70% of sporadic parathyroid carcinomas; encodes parafibromin (transcriptional regulator). Germline CDC73 mutations cause the HPT-JT syndrome (hyperparathyroidism-jaw tumor syndrome).
Familial Syndromes
| Syndrome | Key Features |
|---|---|
| MEN-1 | Parathyroid hyperplasia, pituitary tumors, pancreatic neuroendocrine tumors |
| MEN-2A | Parathyroid hyperplasia, medullary thyroid carcinoma, pheochromocytoma |
| MEN-4 | Similar to MEN-1 |
| FHH (Familial Hypocalciuric Hypercalcemia) | Heterozygous loss-of-function CASR mutations; elevated Ca²⁺ with non-suppressed PTH but NO downstream consequences - does NOT need surgery |
Morphology
Parathyroid Adenoma:
- Almost always solitary
- Average size 0.5-5 g; well-circumscribed, soft, tan-reddish-brown nodule
- Microscopically: sheets of uniform, polygonal chief cells with small central nuclei; nests of larger oxyphil cells also present
- Adipose tissue is inconspicuous (unlike normal parathyroid)
- A rim of compressed non-neoplastic parathyroid tissue at the edge (important diagnostic clue)
- Mitotic figures are rare; bizarre nuclei may be seen (endocrine atypia - NOT a criterion for malignancy)
- The other glands are normal or shrunken due to feedback inhibition
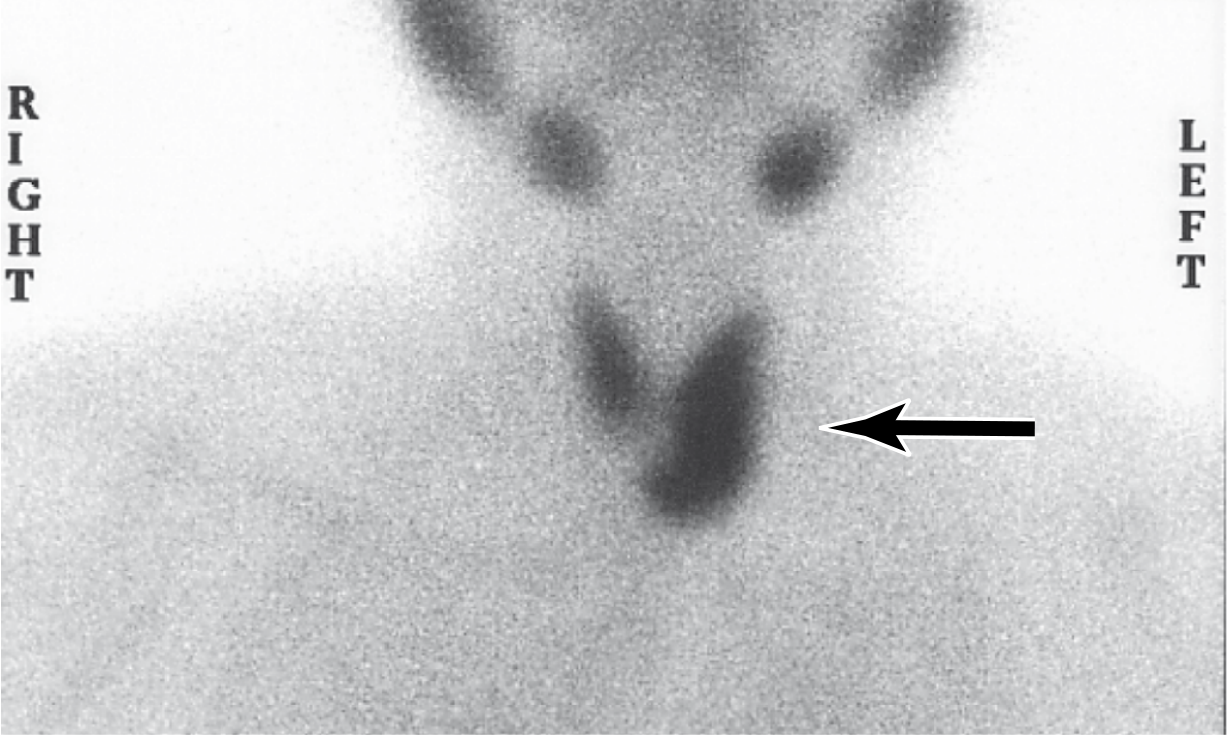
Fig. 24.24 - Tc-99m-sestamibi radionuclide scan showing increased uptake at the left inferior parathyroid (arrow), corresponding to a parathyroid adenoma. Preoperative scintigraphy distinguishes adenomas (single gland uptake) from hyperplasia (multiple glands).
Primary Hyperplasia:
- May be sporadic or part of MEN syndrome
- All four glands involved (though asymmetrically - making it hard to distinguish from adenoma)
- Combined weight rarely exceeds 1 g
- Chief cell hyperplasia most common (diffuse or multinodular); less commonly "water-clear cell hyperplasia"
Parathyroid Carcinoma:
- Defined by invasive growth and/or metastasis (cytology is unreliable)
- Involves single gland; gray-white irregular masses, sometimes >10 g
- Local recurrence in 1/3 of cases; distant metastasis in another 1/3
- Atypical parathyroid neoplasms: new entity; fibrosis, cystic change, macronucleoli without definitive invasion - many show parafibromin loss
Skeletal Changes (Morphology)
Untreated symptomatic 1° HPT leads to three interrelated skeletal abnormalities:
- Osteoporosis - decreased bone mass, preferential involvement of phalanges, vertebrae, proximal femur; cortical bone affected more than medullary bone
- Dissecting osteitis - osteoclasts tunnel along trabeculae creating "railroad track" appearance
- Brown tumors - collections of osteoclasts, reactive giant cells, and hemorrhagic debris that replace normal bone; yellow-brown color (not a true neoplasm); associated with osteitis fibrosa cystica
- Osteitis fibrosa cystica - end-stage: cystic spaces filled with fibrous tissue and hemosiderin-laden macrophages
Renal Changes (Morphology)
- Nephrolithiasis (kidney stones) - most common renal complication; calcium oxalate or calcium phosphate stones
- Nephrocalcinosis - calcium deposition in the renal parenchyma; can impair renal function
Clinical Features - The Classic Mnemonic
"Painful bones, renal stones, abdominal groans, and psychic moans"
| System | Manifestations |
|---|---|
| Skeletal | Bone pain, pathological fractures, osteitis fibrosa cystica, brown tumors |
| Renal | Kidney stones (Ca-oxalate/phosphate), nephrocalcinosis, polyuria, polydipsia |
| GI (abdominal groans) | Nausea, vomiting, constipation, peptic ulcer disease (Ca²⁺ stimulates gastrin), pancreatitis |
| Neuropsychiatric (psychic moans) | Depression, cognitive dysfunction, anxiety, fatigue, weakness |
| Cardiovascular | Hypertension, shortened QT interval |
| Asymptomatic | ~80% - incidentally discovered hypercalcemia |
Lab Findings
- Elevated serum calcium (hypercalcemia)
- Elevated PTH (inappropriately high or frankly elevated)
- Hypophosphatemia (PTH causes phosphaturia)
- Hypercalciuria (increased filtered load overwhelms reabsorption)
- Elevated alkaline phosphatase (if bone disease present)
- Elevated urine cAMP (PTH receptor-mediated)
Differential of Hypercalcemia
Primary HPT is the most common cause of asymptomatic hypercalcemia. Malignancy is the most common cause of symptomatic hypercalcemia. Together they account for nearly 90% of all cases of hypercalcemia.
5. Secondary Hyperparathyroidism (2° HPT)
Causes of the underlying hypocalcemia:
- Chronic renal failure (most common)
- Vitamin D deficiency
- Inadequate dietary calcium
- Steatorrhea (fat malabsorption of vitamin D)
Mechanism in Renal Failure:
- Decreased phosphate excretion → hyperphosphatemia
- Elevated phosphate directly depresses serum calcium → stimulates parathyroid activity
- Loss of renal α1-hydroxylase → reduced active vitamin D → decreased intestinal calcium absorption
- Vitamin D deficiency removes its suppressive effect on parathyroid growth and PTH secretion → compound hypersecretion
Morphology
- All parathyroid glands are hypercellular (asymmetric enlargement possible)
- Increased chief cells or water-clear cells (diffuse or multinodular)
- Fat cells decreased
- Metastatic calcification in lungs, heart, stomach, blood vessels
- Distinction between hyperplasia and adenoma may be impossible histologically
Clinical Features
- Dominated by the underlying chronic renal failure
- Renal osteodystrophy - bony changes that regress with treatment
- Calciphylaxis - vascular calcification causing ischemic skin/organ damage (serious complication)
- Responds to dietary vitamin D supplementation and phosphate binders
6. Tertiary Hyperparathyroidism
- Develops after long-standing secondary HPT
- Parathyroid activity becomes autonomous and excessive
- Results in hypercalcemia (as opposed to normal/low calcium in 2° HPT)
- May require parathyroidectomy
7. Hypoparathyroidism (Brief)
- Most common cause: inadvertent removal or injury during thyroid surgery
- Less common: autoimmune destruction, DiGeorge syndrome (developmental failure of pharyngeal pouches)
- Results in hypocalcemia → tetany, seizures, Chvostek sign, Trousseau sign
- Dental abnormalities in developmental hypocalcemia: hypoplasia, defective enamel, carious teeth
Pseudohypoparathyroidism: End-organ resistance to PTH. PTH levels are normal or elevated, but target tissues do not respond. Caused by G-protein pathway defects. Also causes resistance to TSH and FSH/LH. Presents with hypocalcemia, hyperphosphatemia, and elevated PTH.
Key Concepts Summary (Robbins)
- 1° HPT is the most common cause of asymptomatic hypercalcemia
- Most cases are sporadic parathyroid adenomas (solitary)
- Adenomas are monoclonal; hyperplasia is multiglandular
- Skeletal triad: bone resorption, osteitis fibrosa cystica, brown tumors
- Renal: nephrolithiasis and nephrocalcinosis
- Clinical mnemonic: "painful bones, renal stones, abdominal groans, psychic moans"
- 2° HPT: hyperphosphatemia and low vitamin D in renal failure → compensatory PTH hypersecretion
- Tertiary HPT: autonomous PTH secretion arising from 2° HPT
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 1010-1013
Explain hypo also
Finding Sources
Reading File
I have all the content needed. Here is the full explanation:
Hypoparathyroidism
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease, pp. 1012-1013
Definition
Hypoparathyroidism is a state of PTH deficiency (or end-organ resistance to PTH), resulting in hypocalcemia and hyperphosphatemia. It is far less common than hyperparathyroidism.
Causes / Etiology
1. Surgical (Most Common Acquired Cause)
Inadvertent damage or removal of parathyroid glands during:
- Thyroidectomy (total or near-total) - most common scenario
- Radical neck dissection for malignancy (glands mistaken for lymph nodes)
- Over-resection of parathyroid tissue during treatment of 1° HPT
2. Autoimmune Hypoparathyroidism
- Associated with chronic mucocutaneous candidiasis and primary adrenal insufficiency
- This triad is called Autoimmune Polyglandular Syndrome type 1 (APS-1)
- Caused by mutations in the AIRE gene (autoimmune regulator)
- Typical presentation sequence in childhood:
- Candidiasis (first, early childhood)
- Hypoparathyroidism (several years later)
- Adrenal insufficiency (adolescence)
3. Autosomal Dominant Hypoparathyroidism
- Caused by gain-of-function mutations in the CASR gene (calcium-sensing receptor)
- Heightened calcium sensing suppresses PTH inappropriately → hypocalcemia + hypercalciuria
- Contrast with FHH (heterozygous loss-of-function CASR) which causes hypercalcemia
4. Familial Isolated Hypoparathyroidism (FIH)
- Rare, autosomal dominant or recessive
- AD form: mutation in the PTH gene itself - impairs processing to the active mature hormone
- AR form: loss-of-function mutations in GCM2 (transcription factor essential for parathyroid development)
5. Congenital Absence of Parathyroid Glands
- Associated with thymic aplasia and cardiovascular defects
- Classic association: 22q11 deletion syndrome (DiGeorge syndrome) - failure of 3rd and 4th pharyngeal pouches to develop → absent parathyroids + absent thymus + cardiac defects
Pathophysiology
↓ PTH → ↓ renal calcium reabsorption + ↓ vitamin D activation + ↓ osteoclast activity → Hypocalcemia
↓ PTH → ↓ phosphate excretion → Hyperphosphatemia
High phosphate further binds ionized calcium and deposits in tissues.
Clinical Features
All manifestations stem from hypocalcemia - the severity depends on how low calcium falls and how quickly.
Neuromuscular (Most Prominent)
Tetany - the hallmark of hypocalcemia:
- Circumoral numbness and paresthesias (tingling of lips, hands, feet) - earliest symptoms
- Carpopedal spasm - classic flexion of wrist/fingers
- Laryngospasm - life-threatening
- Generalized seizures
Classic signs:
| Sign | How to Elicit | What Happens |
|---|---|---|
| Chvostek sign | Tap facial nerve anterior to ear | Twitching of eye, mouth, or nose muscles (subclinical tetany) |
| Trousseau sign | Inflate BP cuff on arm above systolic for 3 minutes | Carpal spasm (wrist flexion, finger extension) - more specific |
Neuropsychiatric
- Emotional instability, anxiety, depression
- Confusion, hallucinations
- Frank psychosis
Intracranial
- Basal ganglia calcification (paradoxically, hypocalcemia causes calcification due to elevated phosphate driving calcium phosphate deposition)
- Parkinsonian-like movement disorders
- Increased intracranial pressure → papilledema
Ocular
- Lens calcification and cataract formation
Cardiovascular
- Prolonged QT interval on ECG (as opposed to shortened QT in hypercalcemia)
- Risk of ventricular arrhythmias
Dental (Developmental Hypocalcemia Only)
These changes are highly characteristic and occur when hypocalcemia is present during development:
- Dental hypoplasia
- Failure of eruption
- Defective enamel and root formation
- Abraded, carious teeth
Pseudohypoparathyroidism (PHP)
A separate entity - PTH levels are normal or elevated, but target organs fail to respond.
Mechanism: Genetic defects in the G-protein pathway (Gsα subunit - GNAS mutations) that is shared across multiple hormone receptors.
Result: End-organ resistance to PTH (and also TSH, FSH, LH - all G-protein-coupled receptors)
| Feature | Hypoparathyroidism | Pseudohypoparathyroidism |
|---|---|---|
| PTH level | Low | Normal or High |
| Ca²⁺ | Low | Low |
| Phosphate | High | High |
| Response to exogenous PTH | Normal (urine cAMP rises) | No response |
| TSH | Normal | May be elevated (TSH resistance) |
Clinical: Hypocalcemia + hyperphosphatemia + elevated PTH. TSH resistance is mild. LH/FSH resistance → hypergonadotropic hypogonadism in females.
Quick Comparison Table: Hypo vs. Hyper
| Feature | Hypoparathyroidism | Primary Hyperparathyroidism |
|---|---|---|
| PTH | ↓ | ↑ |
| Serum Ca²⁺ | ↓ (hypocalcemia) | ↑ (hypercalcemia) |
| Serum PO₄ | ↑ (hyperphosphatemia) | ↓ (hypophosphatemia) |
| Urine Ca²⁺ | ↓ | ↑ |
| QT interval | Prolonged | Shortened |
| Key symptom | Tetany, seizures | Stones, bones, groans, moans |
| Bone disease | Osteomalacia (if chronic) | Osteitis fibrosa cystica, brown tumors |
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 1012-1013
This is a shared conversation. Sign in to Orris to start your own chat.