Essay from General Medicine Topic: Nephrotic Syndrome
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Searching PubMed
"nephrotic syndrome" management
Reading File
Loading Image
Loading Image
Loading Image
Reading File
I now have all the material needed for a comprehensive essay. Let me compile it.
Nephrotic Syndrome
Essay from General Medicine
Definition
Nephrotic syndrome is a clinical constellation arising from glomerular damage that causes excessive loss of protein in the urine. It is classically defined by four cardinal features:
- Proteinuria >3.5 g/24 hours (nephrotic-range)
- Hypoalbuminaemia - serum albumin <2.5 g/dL
- Generalised oedema (often anasarca)
- Hyperlipidaemia - total cholesterol usually >180 mg/dL, with lipiduria
Serum creatinine may be variable at presentation.
Pathophysiology
Nephrotic syndrome occurs when there is impairment of glomerular charge and size selectivity, normally maintained by the glomerular basement membrane (GBM), endothelial cells, and epithelial cells (podocytes). The resultant increase in glomerular permeability allows large molecules such as albumin to escape into the urine.
Two complementary mechanisms produce oedema:
1. Underfill theory: Urinary albumin losses and reduced hepatic synthesis lead to hypoalbuminaemia. The fall in plasma oncotic pressure increases fluid flux into the interstitial spaces, activating the renin-angiotensin-aldosterone system (RAAS) and causing secondary renal sodium retention. Patients with minimal change disease typically fall into this category, with contracted plasma volume and stimulated RAAS.
2. Overfill theory: A primary renal salt retention in the distal nephron and thick ascending limb (TAL) leads to sodium and water accumulation independently of oncotic pressure. Patients with other causes of nephrotic syndrome (e.g., membranous nephropathy) often have expanded plasma volume and suppressed RAAS.
Hyperlipidaemia occurs because hypoalbuminaemia stimulates generalised hepatic lipoprotein synthesis, and because reduced plasma oncotic pressure impairs normal lipase activity - raising VLDL and LDL. Lipiduria follows as these lipoproteins leak through the damaged glomerulus, appearing as oval fat bodies and fatty casts in urine.
Clinical Features
Symptoms:
- Peripheral oedema - often starting periorbitally (especially in children), progressing to anasarca
- Frothy (foamy) urine from heavy proteinuria
- Fatigue and dyspnoea (from pleural effusions, ascites)
Physical findings:
- Hypertension (variable - common in secondary causes)
- Generalised dependent pitting oedema, ascites, pleural effusions
- Muehrcke's lines (paired white transverse bands on nails) from hypoalbuminaemia
- Eruptive xanthomata and xanthelasma from hyperlipidaemia
Investigations
Urine:
- 24-hour urine protein (>3.5 g/day confirms nephrotic range); or spot urine protein:creatinine ratio (correlates gram for gram with 24-hour collection - a validated, practical alternative)
- Urine microscopy: oval fat bodies, fatty casts, lipid droplets; may see waxy casts; haematuria is NOT a feature (distinguishes from nephritic syndrome)
- Urine albumin:creatinine ratio
Blood:
- Serum albumin (<2.5 g/dL), total protein
- Fasting lipid profile (elevated total cholesterol, LDL, triglycerides)
- Serum creatinine, urea, electrolytes (GFR estimation)
- Blood glucose/HbA1c (screen for diabetic nephropathy)
- Complement levels (C3, C4) - low in SLE nephritis, MPGN
- ANA, anti-dsDNA (SLE), ANCA, anti-GBM
- HBsAg, HCV antibodies, HIV serology (secondary causes)
- Serum/urine protein electrophoresis (myeloma)
- Anti-PLA2R antibodies (primary membranous nephropathy - positive in ~70%)
Renal biopsy:
Required in most adults (exceptions: typical childhood presentation strongly suggesting minimal change disease, or clear secondary cause such as diabetic nephropathy with characteristic retinopathy). Biopsy guides specific immunosuppressive treatment and provides prognostic information.
Causes
Primary (Idiopathic) Causes
Primary glomerulonephropathies are not related to known systemic disease.
| Subtype | Adults (%) | Key Features |
|---|---|---|
| Membranous nephropathy | 25-35 | Most common in white adults |
| FSGS | 20-25 | Most common in Black patients |
| Minimal change disease | 5-15 | Most common in children |
| MPGN | 5-10 | Mixed nephrotic-nephritic |
| IgA nephropathy | <5 | Usually nephritic |
1. Minimal Change Disease (MCD)
The most frequent cause of nephrotic syndrome in children (1-7 years), responsible for ~80% of childhood cases. Accounts for approximately 5-15% of primary nephrotic syndrome in adults.
Pathogenesis: The leading hypothesis involves circulating molecules - potentially from T cells or other immune cells - that injure podocytes and cause foot process effacement. Recent evidence points to overexpression of angiopoietin-like-4 and CD80 in podocytes. A subset of patients have antibodies to nephrin, a slit-diaphragm protein. The underlying trigger remains incompletely characterised.
Morphology:
- Light microscopy: Glomeruli appear entirely normal (hence the name) - no proliferation, thickening, or deposits
- Immunofluorescence: Negative (no immunoglobulin or complement deposition)
- Electron microscopy: Diffuse effacement (fusion) of podocyte foot processes - the pathognomonic finding
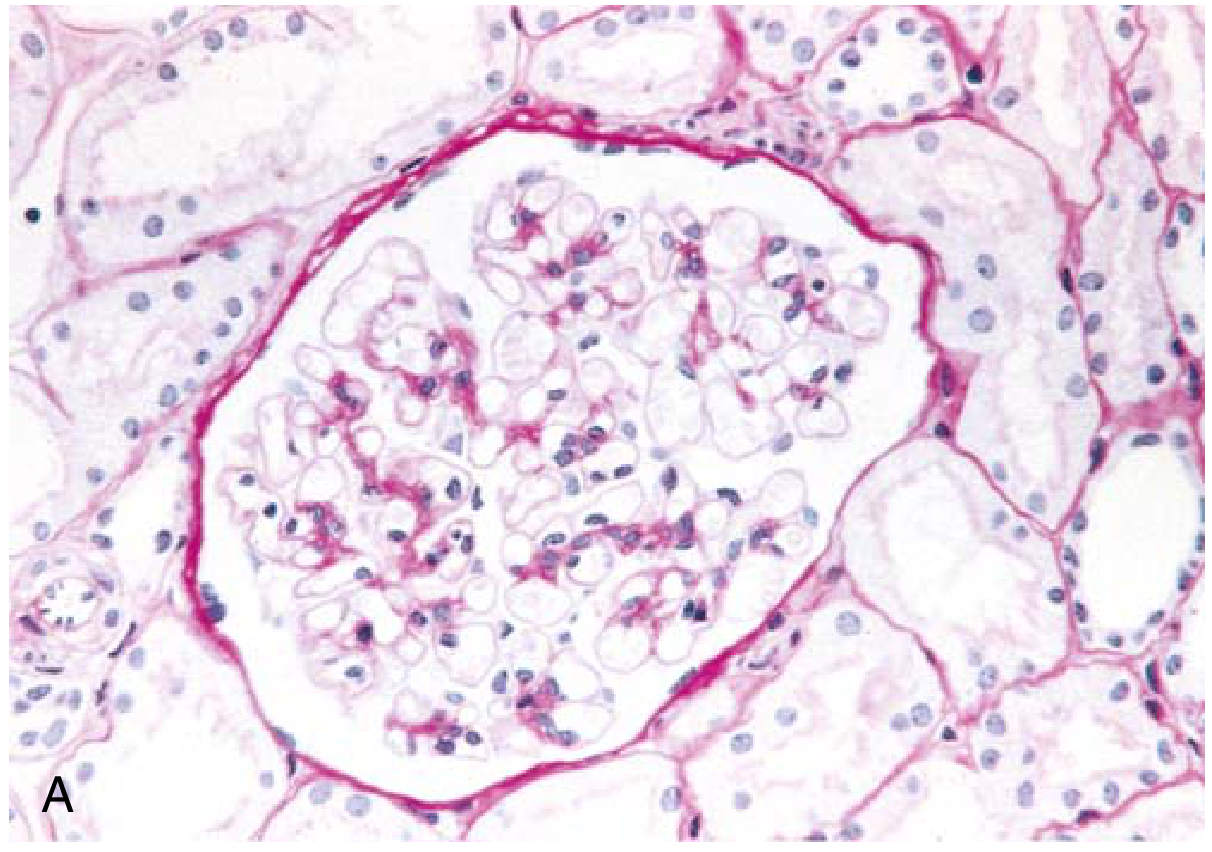
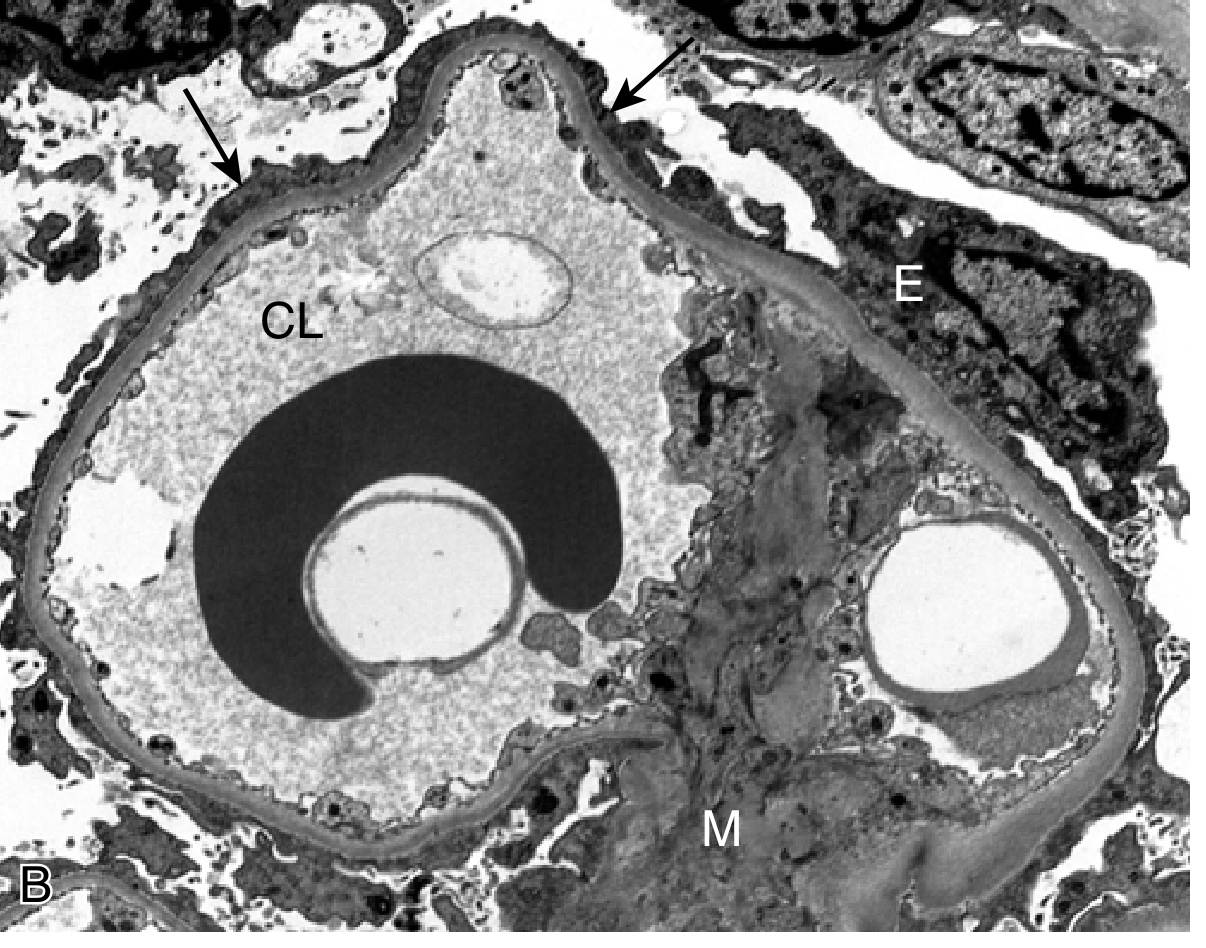
Clinical: Abrupt onset of nephrotic syndrome in an otherwise healthy child. Proteinuria is highly selective (predominantly albumin). Renal function is usually preserved. >90% of children respond to corticosteroids; relapses occur in >2/3. Adults show similar response (>80% remission) but relapses are frequent, sometimes leading to steroid dependence.
2. Focal Segmental Glomerulosclerosis (FSGS)
FSGS accounts for ~30% of all nephrotic syndrome in the United States and is the primary glomerular disorder most commonly progressing to end-stage renal disease. It is most common in Black patients (approximately 50% of glomerular disease cases), with susceptibility linked to genetic polymorphisms in APOL1.
Types:
- Primary FSGS: Likely mediated by a circulating factor (cardiotrophin-like cytokine 1 is one candidate); rapid recurrence after renal transplantation supports this
- Secondary FSGS: Maladaptive response to nephron loss (obesity, remnant kidney), infections (HIV - "HIV nephropathy"), heroin use, genetic mutations in podocyte proteins (nephrin, podocin - >60 genes implicated)
- Collapsing glomerulopathy: A severe variant with collapse of the glomerular tuft and epithelial cell hyperplasia; particularly poor prognosis
Morphology:
- Light microscopy: Focal (some glomeruli affected) and segmental (only part of each glomerulus involved) sclerosis, with obliterated capillary lumina, increased mesangial matrix, hyaline material, and foamy macrophages. Initially affects juxtamedullary glomeruli.
- Immunofluorescence: Nonspecific trapping of IgM and C3 in areas of sclerosis
- Electron microscopy: Diffuse foot process effacement (as in MCD), but with segmental scarring
Clinical: More resistant to steroids than MCD. It carries the highest risk of thromboembolism among primary causes. Progressive renal impairment is common, with up to 50% reaching ESRD within 10 years if untreated.
3. Membranous Nephropathy
The most common cause of primary nephrotic syndrome in white adults. Approximately 70% of cases are primary (anti-PLA2R antibody positive in ~70%), with the remainder secondary to autoimmune disease (SLE), infections (hepatitis B), medications (penicillamine, gold, NSAIDs), or malignancy (colon cancer, lung cancer).
Morphology:
- Light microscopy: Uniform diffuse thickening of the GBM, with "spike and dome" pattern on silver stain (projections of GBM matrix between subepithelial deposits)
- Immunofluorescence: Granular IgG and C3 deposits along the GBM
- Electron microscopy: Subepithelial electron-dense deposits (immune complex deposits)
Clinical: Insidious onset, middle-aged adults. One-third undergo spontaneous remission, one-third remain with persistent proteinuria, one-third progress to renal failure. High risk of renal vein thrombosis.
Secondary Causes
Secondary glomerulonephropathies mirror the clinicopathologic patterns of primary types but arise from identifiable systemic disease or exposures:
| Category | Examples |
|---|---|
| Metabolic | Diabetic nephropathy (Kimmelstiel-Wilson lesions), amyloidosis |
| Autoimmune | SLE (membranous lupus nephritis, Class V), rheumatoid arthritis |
| Infections | HIV, hepatitis B and C, syphilis, malaria (quartan), infective endocarditis, schistosomiasis |
| Drugs/toxins | NSAIDs, lithium, gold, penicillamine, captopril, tamoxifen, heroin |
| Malignancy | Hodgkin's lymphoma (MCD pattern), solid tumours - colon/lung (membranous pattern), multiple myeloma |
| Obstetric | Preeclampsia (resolves post-delivery) |
Diabetic nephropathy: Suspect a non-diabetic cause if nephrotic syndrome occurs with normal renal function, absence of retinopathy, rapid deterioration, active urinary sediment, or short duration of diabetes.
SLE: Can produce both nephrotic (Class V - membranous) and nephritic patterns (Class III/IV - proliferative).
Complications
1. Thromboembolism
The most serious complication. The mechanism involves imbalance between pro- and antithrombotic factors: urinary loss of anticoagulant proteins (antithrombin III, proteins C and S) combined with increased hepatic synthesis of procoagulant proteins (fibrinogen, factor VIII). The most common sites are the renal veins and veins of the lower extremities. Pulmonary embolism can occur. Membranous nephropathy carries the highest thrombotic risk among primary causes.
2. Infection
Nephrotic patients are prone to infections (e.g., spontaneous bacterial peritonitis from ascites, cellulitis, pneumococcal peritonitis) due to:
- Loss of immunoglobulins and complement factors in urine
- Oedema fluid serving as a culture medium
- Immunosuppressive treatment
Encapsulated organisms (especially Streptococcus pneumoniae) are particularly hazardous. Pneumococcal vaccination is recommended.
3. Hyperlipidaemia and Atherosclerosis
Persistent hyperlipidaemia (elevated LDL, VLDL, total cholesterol) significantly increases cardiovascular risk. Statin therapy is warranted in prolonged cases.
4. Acute Kidney Injury
Can arise from: hypovolaemia (especially in underfill MCD), renal vein thrombosis, nephrotoxic drugs (e.g., diuretics, NSAIDs), or intrinsic disease progression.
5. Malnutrition
Sustained protein loss, combined with anorexia and catabolism, leads to muscle wasting and growth retardation in children.
6. Vitamin D Deficiency and Bone Disease
Vitamin D-binding protein is lost in urine, causing hypocalcaemia and secondary hyperparathyroidism.
Management
Management combines general supportive measures with disease-specific immunotherapy guided by renal biopsy findings.
General Measures
Dietary:
- Sodium restriction (<2 g/day) to reduce oedema
- Protein intake: moderate restriction (0.8-1.0 g/kg/day) - high protein intake does not raise serum albumin but worsens proteinuria
Oedema management:
- Loop diuretics (furosemide) are first-line. Note: hypoalbuminaemia reduces furosemide binding to plasma proteins, enlarging its volume of distribution and reducing tubular delivery - higher doses or continuous infusion may be needed
- Addition of a thiazide or potassium-sparing diuretic for synergy in resistant oedema
- Aldosterone antagonists (spironolactone) are useful when hyperaldosteronism is present
Proteinuria reduction:
- ACE inhibitors or ARBs reduce intraglomerular pressure and proteinuria independent of blood pressure. They also combat coagulopathy, dyslipidaemia, oedema, and slow progressive renal function loss. Combination ACE + ARB is generally avoided due to hypotension and worsening renal function risk.
Lipid management:
- Statins for hypercholesterolaemia in patients with prolonged or heavy disease
Anticoagulation:
- Therapeutic anticoagulation (LMWH or warfarin) for confirmed thrombosis (especially renal vein thrombosis or DVT/PE)
- Prophylactic anticoagulation may be considered in membranous nephropathy with very low albumin (<2 g/dL)
Infection prophylaxis:
- Pneumococcal vaccination; consider prophylactic penicillin in highly susceptible children
Disease-Specific Treatment
| Cause | First-Line | Second-Line / Steroid-Resistant |
|---|---|---|
| Minimal change disease | Oral prednisolone (1 mg/kg/day); >80% remission | Cyclosporin A, tacrolimus, mycophenolate for steroid-dependent/resistant cases |
| FSGS (primary) | High-dose corticosteroids (response ~25-50%) | Cyclosporin A, tacrolimus, rituximab (for recurrence after transplant) |
| Membranous nephropathy | Conservative management first (1/3 spontaneous remission); then cyclophosphamide + steroids (Ponticelli regimen) | Rituximab (anti-CD20, effective against PLA2R-positive disease), calcineurin inhibitors |
| Diabetic nephropathy | Strict glycaemic control, RAS blockade (ACEi/ARB), SGLT2 inhibitors | Finerenone (non-steroidal MRA) |
| SLE nephritis (Class V) | Hydroxychloroquine + immunosuppression (mycophenolate/azathioprine) | Belimumab, voclosporin |
Prognosis
Prognosis depends heavily on the underlying cause:
- Minimal change disease: Favourable in children (>90% respond to steroids); frequent relapses but renal failure is uncommon. Adults have a slightly worse response rate but still good long-term outcomes.
- Membranous nephropathy: Variable. One-third remit spontaneously; one-third develop progressive CKD. Anti-PLA2R levels correlate with disease activity and can guide treatment.
- FSGS: The most aggressive primary cause. Up to 50% reach ESRD within 10 years. Collapsing variant has a particularly poor prognosis.
- Secondary causes: Prognosis follows the underlying disease. Preeclampsia resolves post-partum. Diabetic nephropathy progresses to ESRD without aggressive management.
Nephrotic vs. Nephritic Syndrome - Key Distinction
| Feature | Nephrotic | Nephritic |
|---|---|---|
| Proteinuria | >3.5 g/day (massive) | <3.5 g/day (moderate) |
| Haematuria | Absent/minimal | Present (dysmorphic RBCs, RBC casts) |
| Oedema | Prominent, often anasarca | Present but milder |
| Hypertension | Variable | Usually present |
| Renal function | Variable | Often acutely impaired |
| Serum albumin | Low | Normal or slightly reduced |
| Mechanism | Loss of protein selectivity | Inflammatory glomerular damage |
Summary
Nephrotic syndrome is a clinically significant glomerular disorder defined by heavy proteinuria (>3.5 g/day), hypoalbuminaemia, oedema, and hyperlipidaemia. Its causes range from primary podocytopathies (MCD, FSGS, membranous nephropathy) to secondary systemic disorders (diabetes, SLE, infections, malignancies). Pathophysiology centres on podocyte injury leading to loss of glomerular barrier function, with downstream consequences of oedema, thrombophilia, dyslipidaemia, and immune compromise. Renal biopsy is the cornerstone of diagnosis in adults and directs immunosuppressive therapy. Management combines supportive measures (diuretics, RAS blockade, statins, anticoagulation when indicated) with cause-specific treatment. Prognosis is most favourable in MCD and least favourable in FSGS or established diabetic nephropathy.
Sources:
- Frameworks for Internal Medicine (Wilkins & Williams), Chapter 34: Renal
- Robbins & Kumar Basic Pathology, Chapter 12: Kidney
- Brenner and Rector's The Kidney, Chapter 50: Oedema and Diuretics; Chapter 17: Glomerular Disease
- Comprehensive Clinical Nephrology, 7th Edition, Chapter 3: Pathogenesis of Glomerular Syndromes
- Swanson's Family Medicine Review, Chapter on Nephrology
- Goldman-Cecil Medicine, Table 107-2: Subtypes of Idiopathic Nephrotic Syndrome in Adults
- Recent evidence: Interventions for steroid-resistant nephrotic syndrome in children - Cochrane Review, 2025 (PMID: 40337980)
This is a shared conversation. Sign in to Orris to start your own chat.