Thalidomide reason behind disaster
Reading File
Searching the Web
thalidomide disaster mechanism teratogenicity chirality history
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Here is a thorough breakdown of the thalidomide disaster and its underlying reasons:
The Thalidomide Disaster - Why It Happened
Background
Thalidomide was introduced in West Germany in 1957 and rapidly marketed across Europe, Canada, Australia, and elsewhere as a sedative and hypnotic. Because it appeared safer than barbiturates and caused no apparent overdose deaths, it was also widely prescribed to pregnant women for morning sickness relief. Between 1957 and 1962, an estimated ~12,000 infants were born with severe birth defects attributable to the drug.
The Core Reason: Teratogenicity During Organogenesis
The disaster happened because thalidomide is a potent human teratogen - it directly disrupts fetal development without producing obvious harm in the mother.
Critical Window of Exposure
The most important factor was timing. The drug causes harm specifically during the embryonic period of organogenesis (4-8 weeks after fertilization), when tissues and organs are actively forming. For limb development specifically, the critical window is even narrower:
- Exposure 20-36 days after fertilization = severe limb defects
- Earlier in the window = amelia (complete absence of limbs)
- Later in the window = meromelia (partial absence - phocomelia, hypoplasia of radius/ulna)
Women took the drug not realizing they were pregnant or not knowing the risk, and one dose during this narrow window was enough.
"The critical period of limb development is from 24 to 36 days after fertilization. Exposure to this teratogen before day 36 can cause severe limb defects such as amelia."
- The Developing Human: Clinically Oriented Embryology
The Defects Produced
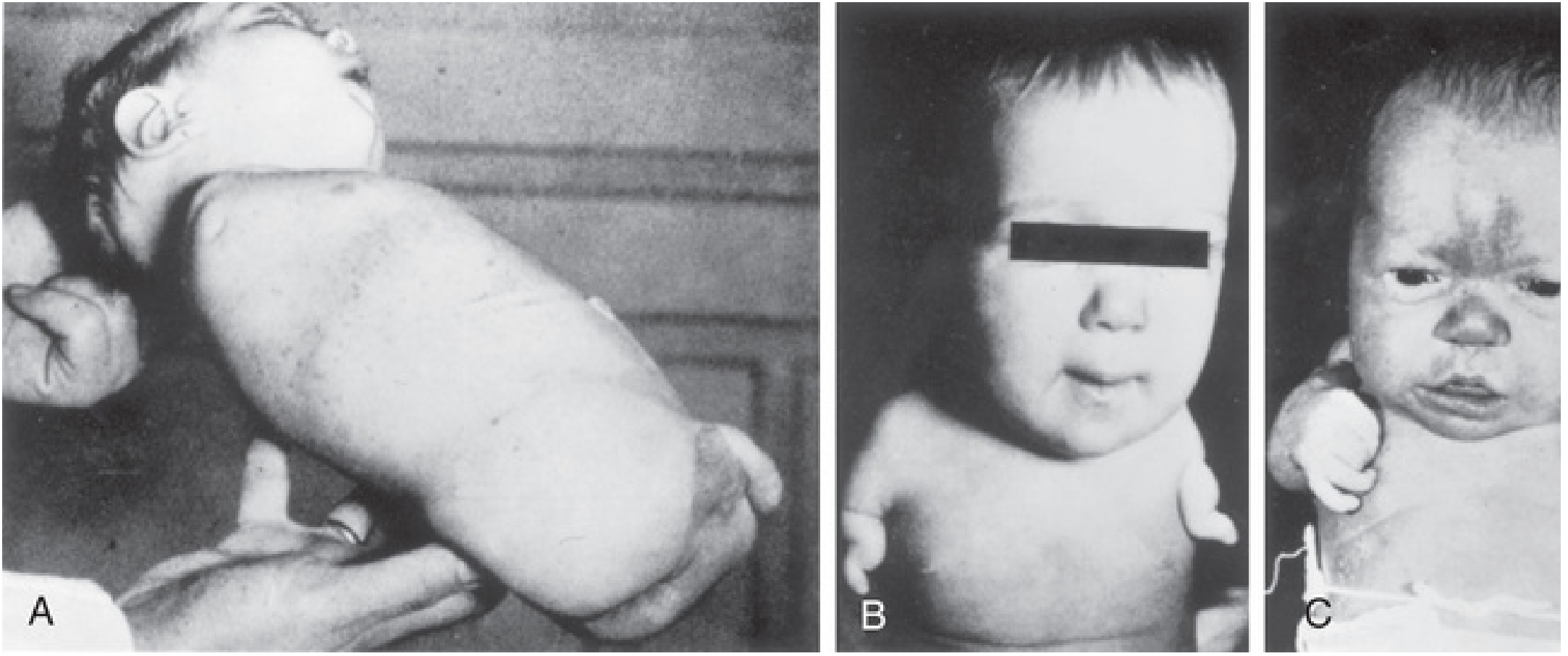
Defects included:
| Defect | Description |
|---|---|
| Amelia | Complete absence of one or more limbs |
| Meromelia / Phocomelia | Hands/feet attached directly to trunk ("seal limbs") |
| Micromelia | Abnormally short/small limbs |
| Ear anomalies | Absence of external/internal ear structures |
| Facial hemangiomas | Vascular birthmarks |
| Cardiac defects | Heart malformations |
| GI/urinary anomalies | Alimentary and urinary system defects |
Molecular Mechanism: Why Thalidomide Is Teratogenic
For decades, over 2,000 papers were written attempting to explain the mechanism, with 15-16 plausible hypotheses. The definitive answer came in 2010:
1. Cereblon (CRBN) Binding - The Primary Mechanism
Thalidomide binds to cereblon (CRBN), a component of a cullin-RING E3 ubiquitin ligase (CRL) complex. When thalidomide binds cereblon, it changes the ligase's substrate specificity, causing it to ubiquitinate and degrade transcription factors (including Ikaros zinc finger family proteins - IKZF1, IKZF3) that are essential for normal embryonic development. Degrading these factors during limb bud development arrests vascular and tissue formation.
2. Anti-Angiogenic Effect
Experimental studies confirm that thalidomide disrupts the formation of early blood vessels in limb buds. Since developing limb buds are entirely dependent on a proper vascular supply, inhibiting angiogenesis at this stage is catastrophic - cells cannot get oxygen or nutrients and the limb fails to form.
The Chirality Problem
Thalidomide has a chiral center (a single stereogenic carbon), existing as two mirror-image enantiomers:
| Enantiomer | Effect |
|---|---|
| (R)-thalidomide | Sedative / therapeutic effect |
| (S)-thalidomide | Teratogenic (embryotoxic) |
The drug was marketed as a racemic mixture (50:50 mix of both forms). A natural thought would be: "Why not just use the pure R-form?"
The answer: in vivo chiral inversion. Even if pure (R)-thalidomide is administered, the body rapidly interconverts the two enantiomers in the liver. The "safe" R-form is converted to the "toxic" S-form bidirectionally. Therefore, separating enantiomers would NOT have prevented the disaster - this is called the "thalidomide paradox."
Why the Disaster Was So Large: Regulatory Failures
Beyond the pharmacology, systemic failures amplified the catastrophe:
- Inadequate pre-market testing - Animal teratogenicity testing on pregnant animals was not required or performed. Early animal studies used non-pregnant models, missing the entire embryotoxic effect.
- Species specificity - Thalidomide's teratogenicity is highly species-selective. Rats and mice showed little harm at standard doses, which gave false reassurance. Rabbits showed the effect only at higher doses. Humans are uniquely sensitive.
- Over-the-counter availability - In many countries thalidomide was sold without prescription, meaning no physician gatekeeping.
- Delayed recognition - The association between thalidomide and birth defects took years to establish. Dr. Frances Kelsey at the US FDA famously blocked its approval in the US, limiting American casualties while demanding more safety data.
- Manufacturers downplayed reports - Early warning signals from physicians were dismissed by the manufacturer (Chemie Grünenthal).
Key Lessons & Legacy
- Thalidomide triggered worldwide overhaul of drug approval regulations, including mandatory teratogenicity testing in multiple animal species before human use.
- In the US, it accelerated passage of the 1962 Kefauver-Harris Amendment to the Food, Drug, and Cosmetic Act.
- Thalidomide itself has since made a comeback - it is now approved for multiple myeloma and erythema nodosum leprosum, with strict risk management programs (REMS in the US) requiring pregnancy tests and contraception.
Sources: The Developing Human: Clinically Oriented Embryology | Thompson & Thompson Genetics and Genomics in Medicine | Goodman & Gilman's Pharmacological Basis of Therapeutics | Goldman-Cecil Medicine
Is there any miss testing in rodent and non rodent
Finding Sources
Searching the Web
thalidomide why missed in rodent testing species differences teratogenicity preclinical
Reading Web Page
https://embryo.asu.edu/pages/studies-thalidomides-effects-ro…
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive information from both the textbook and web sources. Here is a thorough answer:
Why Thalidomide Was Missed in Rodent and Non-Rodent Testing
The Short Answer: Testing Was Either Absent or Fundamentally Flawed
The failure was two-layered: (1) inadequate or absent testing on pregnant animals before market approval, and (2) genuine species-specific biological differences that would have blunted the signal even if testing had been done properly.
Layer 1 - What Testing Actually Happened (or Didn't)
The Disputed Record
There is genuine historical debate about what was tested. Two camps exist:
- Critics claim no testing on pregnant animals was ever done before approval
- Manufacturer's defense claimed some rodent testing was performed and showed no harm
What is clear: no systematic, protocol-driven teratogenicity testing in pregnant animals was conducted before the drug went to market in 1957. Pre-market toxicology at the time was hypothesis-driven, not the structured bioassay testing we have today. Testing in non-pregnant animals (to assess sedation and general toxicity) was done and showed thalidomide to be remarkably safe - which actually boosted confidence in the drug.
The Missed Red Flags in Early Studies
A 1962 UK study by Knapp, Christie, and Seller tested thalidomide in pregnant rats, mice, and rabbits and reported no abnormalities in offspring. However, there was a critical flaw: many animals give birth at night, and animals routinely cannibalize malformed offspring before morning rounds. Technicians arriving the next day saw only normal pups. It was only after investigators performed Caesarean sections and observed animals through the night that malformed pups were found in thalidomide-treated groups. This was a major procedural error that masked the true findings.
Layer 2 - Why Rodents Are Genuinely Poor Models for Thalidomide
Even with perfect methodology, rats and mice would have significantly underestimated the human risk. There are three distinct biological reasons:
1. Metabolic Half-Life Differences (CYP Enzymes)
This is the most important pharmacokinetic reason.
| Species | Plasma Half-Life of Thalidomide | Teratogenic Sensitivity |
|---|---|---|
| Mice | Very short (rapid metabolism) | Extremely low |
| Rats | Short | Very low |
| Rabbits | Intermediate | Moderate (at high doses) |
| Non-human primates | Longer | High |
| Humans | Longest | Highest |
Rodents (especially mice) have high metabolic capacity for thalidomide - they break it down much faster than humans. The drug is cleared before it can act on the embryo. The enzyme responsible for the species difference is CYP3A (cytochrome P450 3A) - humans have CYP3A4/5/7 that metabolize thalidomide differently and may generate teratogenic reactive metabolites, while mice rely on a distinct Cyp3a pathway producing a different metabolite profile (5'-hydroxythalidomide as major product in rodents vs. reactive intermediates in humans).
A 2016 study in Scientific Reports created humanized CYP3A mice (mice with human CYP3A4/5/7 genes replacing mouse Cyp3a genes) and showed that only these mice - not normal mice - developed limb abnormalities when given thalidomide. This directly confirmed that the CYP3A metabolic difference is central to the species gap.
2. Cereblon Binding Differences
Thalidomide's teratogenic mechanism requires binding to cereblon (CRBN) and corrupting its E3 ubiquitin ligase activity. The CRBN protein sequence and its associated binding partners (IKZF family transcription factors) differ between species. Human embryonic CRBN is more sensitive to thalidomide-induced substrate hijacking than rodent CRBN, meaning the molecular target itself responds differently.
3. Embryonic Vascular Sensitivity
Thalidomide inhibits early limb-bud angiogenesis. Rodent embryos form limb-bud vasculature on a slightly different timeline and with different dependence on thalidomide-sensitive pathways than human embryos. The critical window (days 20-36 post-fertilization in humans) has no clean equivalent in the condensed rodent gestational schedule.
Layer 3 - Why Non-Rodent Testing Also Fell Short
Rabbits - the non-rodent species typically used - do show thalidomide teratogenicity, but only at very high doses that were considered pharmacologically irrelevant to human therapeutic doses. The dose-response extrapolation failed because:
- The therapeutic dose range in humans was assumed to be safely below any "effect level" seen in rabbits
- Non-human primates (which are most predictive) were not routinely used in pre-approval testing at the time
- The concept of interspecies dose scaling (adjusting for body weight and metabolic rate) was not established in regulatory science in the 1950s
As the textbook states:
"Animal research has shown that there is a dose-response relationship for teratogens, but the dose used in animals to produce defects is often at levels much higher than typical human exposures. Consequently, animal studies are not readily applicable to human pregnancies."
- The Developing Human: Clinically Oriented Embryology
What Changed After Thalidomide: The Regulatory Legacy
The disaster directly created the modern framework for preclinical testing:
| Reform | Detail |
|---|---|
| Two-species requirement | Testing now mandatory in at least one rodent + one non-rodent species |
| Pregnant animal testing | Specifically required in all three reproductive stages (Segment I, II, III protocols) |
| Gestational stage dosing | Animals must be dosed across all stages of pregnancy, not just one window |
| Multiple dose levels | Low, mid, and high dose groups required to establish dose-response |
| Caesarean section evaluation | Prevents cannibalism artifacts; fetuses examined at correct gestational stages |
| US 1962 Kefauver-Harris Amendment | FDA given authority to require proof of safety before approval |
| UK Medicines Act 1968 | Formalized requirements in Britain |
The Society of Toxicology states plainly: "mice are less sensitive to thalidomide than other species, such as non-human primates and rabbits - as a consequence, developmental toxicity testing for pharmaceuticals now requires assays in at least two species."
Summary Table
| Failure Type | Mechanism | Modern Fix |
|---|---|---|
| No pregnant animal testing | Regulatory gap, no requirement | Mandatory Segment II teratogenicity studies |
| Rodent insensitivity | Short half-life (rapid CYP metabolism), CRBN binding differences | Non-rodent species also required |
| Cannibalism artifacts | Malformed pups eaten before observation | C-section sacrifice, timed necropsies |
| Dose extrapolation failure | Rabbit doses too high vs. human dose | Allometric dose scaling, multiple dose levels |
| Species selection | Only mice/rats used | At least one non-rodent (usually rabbit) required |
| No non-human primate data | Not standard practice | NHP studies for high-risk compounds |
The thalidomide disaster is the founding case study for why modern drug development requires multi-species, multi-stage reproductive toxicity testing before any drug reaches a pregnant human.
Sources: The Developing Human: Clinically Oriented Embryology | Embryo Project Encyclopedia - ASU | Scientific Reports - CYP3A humanized mouse model | Society of Toxicology Impact Statement
Give article on that
Searching PubMed
thalidomide teratogenicity species differences animal testing
Searching PubMed
thalidomide rodent non-rodent preclinical developmental toxicity
Searching PubMed
thalidomide teratogenesis history mechanisms
Searching PubMed
thalidomide CYP3A metabolism species specific teratogenicity
Reading Article
PMID: 26043938
Reading Article
PMID: 26903378
Reading Article
PMID: 30335598
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.