Here are detailed explanations for all 10 clinical case histories from your Physiology Practical-I paper, including symptom analysis, differential diagnosis reasoning, and answers to every question.
PHYSIOLOGY PRACTICAL - I: COMPLETE CASE HISTORY ANSWERS
CASE 1 (Page 1) - HEMOPHILIA
Case Summary
3-year-old male | Bleeding from multiple sites after trivial injury | Joint swelling with pain and fever | Persistent (not profuse) bleeding | Similar history in male relatives | Coagulation time prolonged | Bleeding time, PT, platelet count - ALL NORMAL
Symptom Analysis & Differential Diagnosis Reasoning
Key symptoms and what they tell us:
| Symptom | Significance |
|---|
| Bleeding after trivial injury | Coagulation defect (not vascular) |
| Bleeding from nose, mouth, urinary tract, skin | Multiple sites - systemic coagulopathy |
| Hemarthrosis (joint swelling, pain, fever) | Classic for factor deficiency disorders |
| Persistent, not profuse bleeding | Secondary hemostasis defect (clot doesn't form) |
| Only males affected in family | X-linked inheritance |
| Prolonged coagulation time | Intrinsic pathway defect |
| Normal bleeding time | Primary hemostasis (platelets + vessels) intact |
| Normal PT | Extrinsic pathway (Factor VII) intact |
| Normal platelet count | Not thrombocytopenia |
How to rule out other diseases:
- Von Willebrand Disease - ruled out: vWD prolongs both bleeding time AND coagulation time (since vWF assists platelet adhesion). Here bleeding time is normal.
- Thrombocytopenia (ITP) - ruled out: platelet count is normal; ITP causes petechiae and prolonged bleeding time.
- Vitamin K deficiency - ruled out: this prolongs PT (extrinsic + final common pathway). Here PT is normal.
- Liver disease - ruled out: liver disease prolongs PT and multiple clotting factors; PT is normal here.
- DIC - ruled out: DIC consumes platelets; platelet count is normal.
The combination of prolonged coagulation time (intrinsic pathway) + normal PT + normal platelets + normal bleeding time + X-linked family history + hemarthrosis points exclusively to Hemophilia A (Factor VIII deficiency) or Hemophilia B (Factor IX deficiency).
Answers
Q1. Most likely diagnosis:
Hemophilia A (Deficiency of Factor VIII). Hemophilia B (Christmas disease - Factor IX deficiency) is also possible but Hemophilia A is 4x more common. Both present identically clinically.
The prolonged coagulation time (whole blood clotting time) reflects a defect in the intrinsic coagulation pathway (Factors XII → XI → IX → VIII → X). Factor VIII/IX are part of this pathway. PT tests the extrinsic + common pathway (Factor VII → X → V → II → I), which is intact here. Bleeding time tests platelet function and vascular response - also intact.
Q2. Why are females usually not affected?
Hemophilia is an X-linked recessive disorder. The gene for Factor VIII (Hemophilia A) is located on the X chromosome.
- Males are hemizygous (XY): if they inherit the defective X, they have no second X to compensate → disease manifests.
- Females are heterozygous carriers (X^H X): the normal X chromosome produces enough Factor VIII to prevent bleeding. They are carriers but clinically unaffected.
- For a female to be affected, she would need to be homozygous (X^H X^H), which is extremely rare (would require a carrier mother AND hemophiliac father).
Q3. Chances of the boy's sister being a carrier:
The boy's mother is likely a carrier (X^H X). When a carrier mother has children:
- Each daughter has a 50% chance of being a carrier (X^H X) and 50% chance of being normal (X X).
- Therefore, the probability of the boy's sister being a carrier = 50% (1 in 2).
Q4. Physiological basis of treatment:
The treatment is replacement therapy - giving the missing clotting factor.
- Hemophilia A: Infusion of Factor VIII concentrate (recombinant or plasma-derived). Fresh frozen plasma (FFP) or cryoprecipitate (rich in Factor VIII, vWF, fibrinogen) can also be used.
- Hemophilia B: Infusion of Factor IX concentrate or FFP.
- Physiological basis: Replacing the deficient factor restores the intrinsic coagulation cascade, allowing thrombin generation, fibrin clot formation, and arrest of bleeding.
- Adjunct: Desmopressin (DDAVP) in mild Hemophilia A - stimulates release of stored vWF and Factor VIII from endothelium.
CASE 2 (Page 2) - IRON DEFICIENCY ANEMIA
Case Summary
30-year-old woman | Fatigue, breathlessness, giddiness, headache, palpitation, anorexia, dysphagia | Pallor, tachycardia, glossitis, koilonychia (spoon nails) | Tingling in fingers/toes, dependent edema | Hb: 6 g% | RBC: 3 million/mm³ | MCHC: 28 pg (low) | MCV: 60 µ³ (low) | Eosinophilia | Hookworm ova in stool
Symptom Analysis & Differential Diagnosis Reasoning
Symptoms explained:
| Symptom | Mechanism |
|---|
| Fatigue, breathlessness, giddiness, headache | Reduced oxygen-carrying capacity (Hb 6 g%) → tissue hypoxia |
| Palpitation, tachycardia | Compensatory - heart pumps faster to maintain cardiac output despite low Hb |
| Pallor | Reduced Hb content of blood |
| Glossitis | Mucosal atrophy due to iron deficiency (iron needed for cell proliferation) |
| Koilonychia (spoon nails) | Classic sign - iron deficiency weakens nail plate; it concaves |
| Dysphagia | Paterson-Kelly/Plummer-Vinson syndrome - mucosal atrophy with esophageal web |
| Tingling (paresthesia) | Mild peripheral neuropathy from B12/folate co-deficiency (hookworm may cause multiple deficiencies) |
| Dependent edema | Severe hypoproteinemia from malnutrition; also hookworm blood loss → hypoalbuminemia |
| Anorexia | Non-specific; also from hookworm infestation |
Blood film interpretation:
- Hb 6 g% (normal ~12-14 in women) → severe anemia
- MCV 60 µ³ (normal 80-100) → microcytic
- MCHC 28 pg (normal 27-33 pg per cell, but MCHC normal 32-36 g/dL) → hypochromic
- Together: Microcytic hypochromic anemia
- Eosinophilia → parasitic infestation (hookworm = tissue invasion → eosinophilia)
How to rule out other microcytic anemias:
- Thalassemia - also microcytic/hypochromic but: family history present, target cells on smear, normal/elevated ferritin, HbA2 elevated on electrophoresis. No hookworm.
- Anemia of chronic disease - normocytic usually; serum ferritin elevated, TIBC low. No hookworm.
- Sideroblastic anemia - ring sideroblasts on bone marrow; different cause.
- B12/Folate deficiency - megaloblastic (macrocytic, MCV >100), hypersegmented neutrophils. Not microcytic.
The presence of hookworm ova in stool + microcytic hypochromic anemia + koilonychia + glossitis + dysphagia = Iron Deficiency Anemia secondary to hookworm infestation.
Answers
Q1. Type of anemia:
Microcytic Hypochromic Anemia - specifically Iron Deficiency Anemia (IDA).
- MCV 60 µ³ = microcytic (cells are smaller than normal)
- MCHC 28 pg = hypochromic (cells have less hemoglobin, appear pale)
- This is the most common type of anemia worldwide.
Q2. Cause of anemia:
Hookworm infestation (Ancylostoma duodenale / Necator americanus) causing chronic blood loss.
- Each hookworm attaches to the intestinal mucosa and ingests blood (0.2-0.5 mL/worm/day).
- Massive infestation leads to significant chronic iron loss.
- Iron is required for heme synthesis (protoporphyrin + Fe²⁺ + globin → hemoglobin). Deficiency → reduced Hb production → smaller, paler red cells.
- Additionally, hookworm larvae penetrate skin, migrate through lungs (causing eosinophilia), and settle in the gut, causing protein malnutrition as well.
Q3. Principle of treatment:
Three components:
- Iron replacement: Oral ferrous sulfate (most common) - replenishes iron stores and allows resumption of hemoglobin synthesis. Parenteral iron (iron sucrose, ferric carboxymaltose) if oral not tolerated.
- Anti-helminthic therapy: Albendazole or Mebendazole to eliminate hookworm - removes the cause of chronic blood loss.
- Dietary advice: High-iron diet (leafy vegetables, meat, pulses), Vitamin C (enhances non-heme iron absorption by reducing Fe³⁺ → Fe²⁺), avoid tea/coffee with meals (tannins inhibit iron absorption).
CASE 3 (Page 3) - MYASTHENIA GRAVIS
Case Summary
Young female | Abnormal muscle fatigue | Strength initially normal but fatigues with repetition/as day progresses | Ptosis, weakness of chewing/swallowing/speaking | Remitting course, precipitated by emotions/infections | Nervous system examination: normal | Dramatic recovery after intramuscular neostigmine
Symptom Analysis & Differential Diagnosis Reasoning
Pathophysiological reasoning of symptoms:
| Symptom | Mechanism |
|---|
| Fatigue that worsens with use | Depletion of available ACh quanta at NMJ; autoantibodies reduce functional nAChR |
| Ptosis (drooping eyelid) | Weakness of levator palpebrae superioris (neuromuscular junction in ocular muscles affected early) |
| Weakness of chewing/swallowing/speech | Bulbar muscles innervated by cranial nerves have high-frequency firing demand → fatigue faster |
| Normal nervous system exam | The lesion is at the neuromuscular junction (peripheral), not the nerve or CNS |
| Remissions and relapses | Autoimmune disease - fluctuating antibody titers and T-cell activity |
| Precipitated by emotions/infections | Stress → catecholamines compete at NMJ; infections → immune activation → more antibodies |
| Recovery after neostigmine | Neostigmine inhibits acetylcholinesterase → more ACh available at NMJ → improves transmission |
How to rule out other diseases:
- Lambert-Eaton Syndrome - paraneoplastic (usually lung cancer); weakness improves with repetition (opposite pattern); affects proximal limbs not ocular/bulbar muscles primarily. Anti-VGCC antibodies (pre-synaptic).
- Multiple Sclerosis - CNS demyelination; neurological examination would be abnormal; no fatigability pattern.
- Botulism - blocks ACh release (pre-synaptic); descending paralysis; no response to neostigmine (because the problem is no ACh being released, not reduced receptors).
- ALS (Motor Neuron Disease) - upper AND lower motor neuron signs; progressive; no response to neostigmine.
- Muscular Dystrophy - primary muscle disease; not fatigable; no response to neostigmine.
The neostigmine test (Tensilon/edrophonium test in clinical practice) is both diagnostic AND therapeutic confirmation.
Answers
Q1. Diagnosis:
Myasthenia Gravis (MG) - an autoimmune neuromuscular junction disorder.
Q2. Cause of this condition:
Myasthenia Gravis is caused by autoantibodies against nicotinic acetylcholine receptors (nAChR) at the motor end plate (post-synaptic membrane of the neuromuscular junction).
- In ~85% of cases: Anti-AChR antibodies (IgG) bind to and destroy or block nAChR → fewer functional receptors → insufficient end-plate potential → muscle cannot reach action potential threshold.
- In ~5%: Anti-MuSK antibodies (muscle-specific kinase - essential for clustering AChR).
- Associated with thymic abnormalities: thymoma (~15%) or thymic hyperplasia (~65%).
- The antibodies are formed by autoreactive B cells; T-cell mediated mechanisms also contribute.
- Because acetylcholine is released normally but fewer receptors are available, with sustained activity, the EPP falls below threshold → fatigue.
Q3. How does neostigmine improve the condition?
Neostigmine is an anticholinesterase (acetylcholinesterase inhibitor).
- Normally, after ACh is released into the synapse, acetylcholinesterase (AChE) rapidly breaks it down into choline + acetate.
- In MG, since fewer receptors are available, even normal ACh release is insufficient.
- Neostigmine inhibits AChE → ACh is NOT broken down → accumulates in the synapse → remains available longer → has more chances to bind the remaining functional receptors → End-plate potential reaches threshold → muscle contracts.
- This does NOT cure MG but compensates for the receptor deficiency by increasing available ACh.
- Pyridostigmine (longer-acting, oral) is the mainstay symptomatic treatment.
CASE 4 (Pages 4-5) - Clinical Charts (Not case histories)
(Page 4 is about a clinical chart/graph - likely a spirometry or ECG. Page 5 involves brain waves. These are chart-based questions without a patient history, so they fall outside the "case history" scope of this answer set. Covered briefly below.)
Page 4 chart - likely a Spirogram or Blood pressure chart:
- a. Name the clinical condition: Based on characteristic lines - likely Cheyne-Stokes respiration or pulsus paradoxus - requires viewing the actual chart.
- The question structure (characteristic lines, causes, 2 lab tests) suggests it may be a Spirometry chart for obstructive/restrictive lung disease.
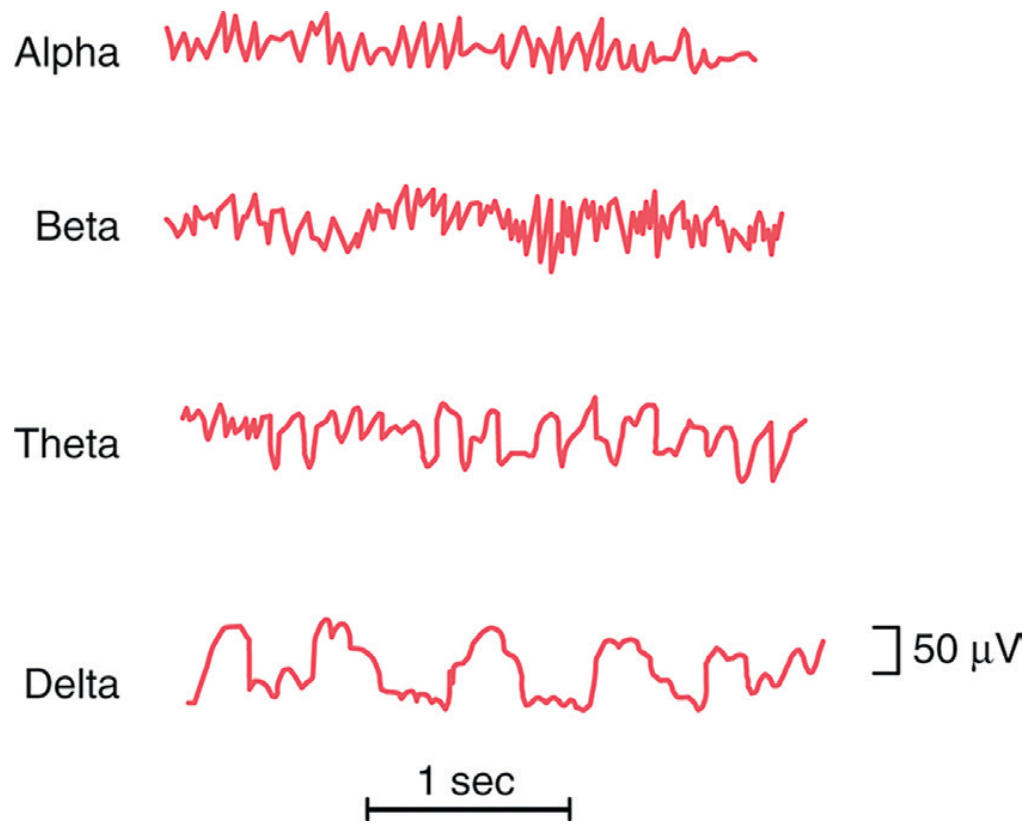
Page 5 chart - EEG (Electroencephalogram) waves:
- a. Waves A, B, C, D = Alpha (α), Beta (β), Theta (θ), Delta (δ) waves
- b. Alpha recorded best: posterior occipital region at rest with eyes closed. Beta: frontal lobes. Theta: temporal/frontal in drowsiness. Delta: deep sleep, recorded over all scalp.
- c. Features: Alpha 8-13 Hz; Beta 13-30 Hz; Theta 4-7 Hz; Delta 0.5-4 Hz.
- d. Clinical uses: Epilepsy diagnosis, sleep disorders, coma grading, brain death, encephalopathy, monitoring anesthesia depth.
CASE 8 (Page 8) - HYPERTHYROIDISM (GRAVES' DISEASE)
Case Summary
28-year-old female | Nervousness, restlessness, tiredness, excessive sweating, palpitation | Increased appetite, amenorrhea | Tachycardia, dyspnea on exertion, fine tremors of outstretched hands, eyelid retraction | BMR: +40% | Basal pulse rate: 120/min
Symptom Analysis & Differential Diagnosis Reasoning
Thyroid hormone excess explains every symptom:
| Symptom | Mechanism |
|---|
| Nervousness/restlessness | Increased CNS excitability; thyroid hormones increase adrenergic sensitivity |
| Excessive sweating | Increased BMR → increased heat production → sweating to dissipate heat |
| Weight loss despite increased appetite | Thyroid hormones increase catabolism (protein + fat breakdown) faster than food intake can compensate |
| Palpitation/tachycardia (120/min) | T3/T4 increase cardiac β1-adrenergic sensitivity → increased heart rate and contractility |
| Dyspnea on exertion | High cardiac output increases oxygen demand; respiratory muscles also weakened |
| Fine tremors | Increased neuromuscular excitability; muscle protein catabolism |
| Eyelid retraction | Sympathetic overactivity → superior tarsal muscle (Müller's muscle) contracts → lid retraction (not true proptosis unless Graves' ophthalmopathy) |
| Amenorrhea | Thyroid hormones disrupt HPO axis; increased SHBG affects estrogen metabolism |
| Tiredness | Despite high BMR, muscle wasting → actual weakness |
BMR +40% (normal: ±15%): Thyroid hormones are the primary regulators of BMR via uncoupling oxidative phosphorylation and upregulating Na⁺/K⁺-ATPase.
How to rule out other conditions:
- Anxiety disorder/panic disorder - tachycardia and nervousness overlap, but no weight loss, no raised BMR, no eye signs, TSH normal.
- Phaeochromocytoma - also sweating, tachycardia, hypertension but episodic; BMR mildly elevated; urine catecholamines elevated; TSH normal.
- Diabetes - weight loss + increased appetite but no tachycardia, no tremors, no eye signs; blood glucose elevated.
Answers
Q1. Comment on investigation report:
- BMR +40%: Significantly elevated (normal is ±15%). This indicates a hypermetabolic state. Thyroid hormones increase the rate of oxidative phosphorylation in all cells by increasing expression of mitochondrial enzymes and Na⁺/K⁺-ATPase. Every 1% rise in BMR correlates with approximately 1% increase in oxygen consumption. +40% is markedly hypermetabolic, consistent with severe hyperthyroidism.
- Basal pulse rate 120/min: Significantly elevated (normal 60-100/min). Persistent resting tachycardia reflects increased cardiac sympathetic tone due to thyroid hormone-induced upregulation of β-adrenergic receptors. This explains palpitations and dyspnea.
Q2. Diagnosis:
Hyperthyroidism - most likely Graves' Disease (autoimmune), given:
- Young woman (most common in females 20-40 years)
- Diffuse features (not nodular)
- Eyelid retraction (sympathetic manifestation)
- Classic constellation of symptoms
Q3. Other investigations to confirm diagnosis:
- Serum TSH (Thyroid Stimulating Hormone): Will be extremely LOW (suppressed) in hyperthyroidism due to negative feedback on anterior pituitary. This is the most sensitive screening test.
- Serum Free T3 and Free T4 levels: Will be markedly elevated - directly confirms hyperthyroidism.
- Thyroid Antibodies: Anti-TSH Receptor antibodies (TRAb/TSHR-Ab) - positive in Graves' disease (stimulating antibodies activate TSH receptors). Also anti-TPO and anti-thyroglobulin antibodies.
- Radioactive Iodine Uptake (RAIU) scan: Diffusely elevated uptake in Graves'; focal uptake in toxic nodule.
- Thyroid ultrasound: Diffuse enlargement with increased vascularity in Graves' disease.
CASE 9 (Page 9) - DIABETES MELLITUS TYPE 2
Case Summary
Middle-aged individual | Weakness, increased thirst (polydipsia), increased urine output (polyuria), increased appetite (polyphagia) | Weight loss | Poor wound healing | Urine: sugar present, NO ketone bodies | Fasting blood glucose: 160 mg/dl
Symptom Analysis & Differential Diagnosis Reasoning
The "3 Ps" of diabetes explained:
| Symptom | Mechanism |
|---|
| Polyuria | High glucose in blood → exceeds renal threshold (~180 mg/dl) → glucose spills into urine → osmotic diuresis → large urine volumes |
| Polydipsia | Osmotic diuresis causes dehydration → hyperosmolarity stimulates hypothalamic thirst center |
| Polyphagia | Glucose cannot enter insulin-sensitive cells (muscle, fat) → cells "starve" despite high blood glucose → hunger signals persist |
| Weight loss | Cells cannot use glucose → body breaks down fat and protein for energy → weight loss despite eating |
| Weakness | Reduced intracellular glucose → reduced ATP production → weakness |
| Poor wound healing | High glucose impairs neutrophil chemotaxis, phagocytosis, and oxidative burst; also damages small vessels (microangiopathy) reducing blood supply to wounds; glycosylated collagen is abnormal |
Fasting glucose 160 mg/dl: Normal fasting glucose is 70-100 mg/dl. 100-125 = impaired fasting glucose (pre-diabetes). ≥126 mg/dl on 2 occasions = Diabetes Mellitus diagnosis. 160 mg/dl is clearly diagnostic.
How to rule out other conditions:
- Diabetes Insipidus - polyuria + polydipsia but NO glycosuria, NO elevated blood glucose. Urine is very dilute (low osmolarity). Due to ADH deficiency (central DI) or resistance (nephrogenic DI).
- Type 1 DM - would have ketone bodies in urine (absolute insulin deficiency → unrestrained lipolysis → ketogenesis). Middle age, gradual onset, and absence of ketones suggests Type 2.
- LADA (Latent Autoimmune Diabetes of Adults) - possible but less likely without autoantibody testing.
Answers
Q1. Diagnosis:
Type 2 Diabetes Mellitus - based on:
- Classic triad: polyuria, polydipsia, polyphagia
- Weight loss and poor wound healing
- Glucosuria (sugar in urine)
- Fasting blood glucose 160 mg/dl (>126 mg/dl = DM)
- Absence of ketone bodies - key finding suggesting Type 2, not Type 1
Q2. Causes for the presenting symptoms:
Polyuria: When blood glucose exceeds the renal threshold for reabsorption (~180 mg/dl), glucose appears in the filtrate and acts as an osmotic agent, drawing water with it → large volume urine output.
Polydipsia: Osmotic diuresis leads to dehydration and increased plasma osmolality → stimulates hypothalamic osmoreceptors → thirst sensation.
Polyphagia: In Type 2 DM, there is relative insulin deficiency or peripheral insulin resistance. Glucose cannot enter insulin-dependent cells (skeletal muscle, adipose tissue) → intracellular energy deficit → hypothalamus interprets this as starvation → hunger signals (ghrelin elevated, leptin reduced perception) → increased appetite.
Weight loss: Despite adequate caloric intake, cells cannot metabolize glucose → body activates gluconeogenesis (breaking down muscle protein) and lipolysis (breaking down fat) → progressive weight loss.
Poor wound healing:
- Hyperglycemia impairs neutrophil and macrophage function (chemotaxis, phagocytosis)
- Glycosylation of proteins (advanced glycation end-products) damages blood vessel walls → microangiopathy → reduced local blood flow to wounds
- Reduced collagen synthesis and cross-linking
- Peripheral neuropathy (in chronic DM) reduces sensation → injuries go unnoticed
Q3. What does absence of ketone bodies suggest?
Absence of ketone bodies suggests Type 2 Diabetes Mellitus (not Type 1):
- In Type 1 DM, there is absolute insulin deficiency. Without any insulin, adipose tissue undergoes unrestrained lipolysis → large amounts of free fatty acids flood the liver → beta-oxidation overwhelms the TCA cycle → acetyl-CoA is diverted to ketone body synthesis (acetoacetate, beta-hydroxybutyrate, acetone) → ketonuria and diabetic ketoacidosis (DKA).
- In Type 2 DM, there is relative insulin deficiency or insulin resistance. Even small amounts of residual insulin are sufficient to suppress lipolysis in adipose tissue → free fatty acid flux to liver is NOT excessive → ketone body synthesis is not significantly elevated → urine is ketone-free.
- This is also why Type 2 DM rarely develops DKA spontaneously (though it can in extreme stress).
CASE 10 (Page 10) - PEPTIC ULCER (DUODENAL)
Case Summary
35-year-old business executive | Upper abdominal pain relieved by food | Basal HCl secretion: 6 mEq/L | Augmented histamine test secretion: 35 mEq/L
Symptom Analysis & Differential Diagnosis Reasoning
The pain relieved by food is the pathognomonic clue here. This points to a duodenal ulcer rather than gastric ulcer (in gastric ulcer, food often worsens pain because it stimulates more acid against the ulcerated gastric wall).
Acid secretion values:
- Normal basal secretion: 0-5 mEq/hr (this patient = 6 mEq/L - elevated)
- Augmented histamine test (maximal acid output): normal = 20-30 mEq/hr (this patient = 35 mEq/L - significantly elevated)
Elevated basal and maximal acid output suggests hyperacidity, consistent with duodenal ulcer.
How to rule out other conditions:
- Gastric ulcer - pain worsened or not relieved by food; often in older patients; weight loss from food aversion.
- Gastric carcinoma - progressive, not relieved by food, weight loss, older age, endoscopy would show mass.
- GERD - burning retrosternal, worse after meals and lying down; not relieved by food per se.
- Pancreatitis - epigastric pain radiating to back, not relieved by food; elevated amylase/lipase.
Answers
Q1. Likely diagnosis:
Duodenal Peptic Ulcer (most likely).
- Pain in the upper abdomen (epigastric), relieved by food, is classic for duodenal ulcer
- Elevated basal and stimulated HCl secretion supports hyperacidity
- Age and presentation (business executive - stress is a contributing factor) fits
Q2. Normal value for augmented histamine test:
- Normal Maximal Acid Output (MAO) in the augmented histamine test = 20-30 mEq/hour (some sources state up to 25 mEq/hr)
- The test involves subcutaneous injection of histamine (0.04 mg/kg body weight) after antihistamine pre-medication (to block systemic effects but not gastric H2 receptors)
- This patient's 35 mEq/L is above normal, indicating hyperchlorhydria (excess acid secretion), characteristic of duodenal ulcer
Q3. Other agent to provoke gastric secretion:
- Pentagastrin (synthetic gastrin analogue) - now the preferred agent as it is safer than histamine (no systemic side effects). Dose: 6 µg/kg subcutaneously.
- Insulin (Hollander insulin test) - causes hypoglycemia → stimulates vagus nerve → stimulates gastric HCl secretion. Used to test completeness of vagotomy.
- Caffeine - stimulates gastric acid secretion via CNS and directly.
Q4. Why is pain relieved by food intake?
The mechanism is acid neutralization and buffering by food:
- In a duodenal ulcer, the ulcer is in the duodenum, just distal to the pylorus. Gastric acid pours into the duodenum and irritates the exposed mucosal surface (where the ulcer is) → pain.
- When food is ingested: Food enters the stomach → acts as a physical and chemical buffer for HCl in the stomach. Proteins in food have strong acid-buffering capacity (amino acid side chains act as buffers).
- Additionally, food in the stomach stimulates pyloric closure and duodenal hormones (secretin, CCK) which temporarily reduce gastric emptying and reduce acid delivery to the duodenum.
- Result: Less free acid reaching the duodenal ulcer → temporary relief of pain.
- Note: Pain returns 2-3 hours after eating once food is digested and buffering capacity is exhausted (this is why duodenal ulcer causes "hunger pain" or "pain at night" - called Moynihan's rhythmicity).
CASE 11 (Page 11) - OBSTRUCTIVE JAUNDICE
Case Summary
Patient | Yellow sclera and skin (jaundice) | Clay-colored (pale), bulky, foul-smelling stool | Itching (pruritus), anorexia | Bradycardia on examination | Stercobilinogen absent in stool | Bilirubin present in urine | Prolonged coagulation | Serum albumin lowered | Serum bilirubin: 6 mg/dl | LFT enzymes elevated | Van den Bergh test: DIRECT POSITIVE
Symptom Analysis & Differential Diagnosis Reasoning
The three types of jaundice and how to differentiate:
| Parameter | Pre-hepatic (Hemolytic) | Hepatic (Hepatocellular) | Post-hepatic (Obstructive) |
|---|
| Urine bilirubin | Absent | Present | Present (++) |
| Urobilinogen | Very high | Variable | Absent/low |
| Stercobilinogen | Very high | Variable | Absent |
| Stool color | Dark | Variable | Clay/pale |
| Van den Bergh | Indirect+ | Both+ | Direct+ |
| Itch | Absent | Mild | Marked |
| Bradycardia | No | No | Yes (bile acids) |
| Coagulation | Normal | Prolonged | Prolonged |
| Fat malabsorption | No | Mild | Yes (bulky, steatorrhea) |
This case: All features point to Post-hepatic (Obstructive) Jaundice (bile duct obstruction - e.g., gallstone in common bile duct, carcinoma of head of pancreas, cholangiocarcinoma).
Answers
Q1. Diagnosis:
Obstructive Jaundice (Post-hepatic / Cholestatic Jaundice) - due to obstruction of the common bile duct or biliary tree.
The complete triad is supported by:
- Direct bilirubin elevation (Van den Bergh direct+): Conjugated bilirubin dams back into blood (it is water-soluble → enters urine)
- Absent stercobilinogen in stool: No bile reaching intestine → no urobilinogen/stercobilinogen produced → clay-colored stool
- Bilirubin in urine (bilirubinuria): Water-soluble conjugated bilirubin filtered by kidneys
- Pruritus: Bile salts deposited in skin stimulate cutaneous nerve endings
- Bradycardia: Bile acids accumulated in blood have a direct negative chronotropic effect on the SA node
- Prolonged coagulation: Bile is essential for absorption of fat-soluble vitamins (A, D, E, K). Obstruction → no bile in gut → Vitamin K malabsorption → reduced synthesis of clotting factors II, VII, IX, X → prolonged PT/coagulation time
- Bulky, foul-smelling stool: Fat malabsorption (steatorrhea) - bile acids needed for fat micelle formation; without bile, fat passes undigested → bulky, greasy, foul stools
- Low serum albumin: Prolonged obstruction → liver damage → reduced albumin synthesis
- Elevated liver enzymes: Backpressure of bile damages hepatocytes → enzyme release
Q2. Why stool was pale, bulky, and foul smelling?
- Pale/clay color: Normally, conjugated bilirubin enters the duodenum via bile duct → intestinal bacteria convert it to urobilinogen → further reduced to stercobilinogen → oxidized to stercobilin → gives stool its normal brown color. In obstruction, NO bile reaches the intestine → NO stercobilinogen → NO stercobilin → stool is pale/clay-colored.
- Bulky: Bile salts are essential for emulsification and absorption of dietary fats. Without bile, fats pass undigested → steatorrhea (fat-laden bulky stools).
- Foul smelling: Undigested and putrefied fat in the intestine undergoes bacterial action → fermentation of unabsorbed organic compounds → offensive odor.
Q3. Why is Van den Bergh test direct positive?
- The Van den Bergh test detects bilirubin in serum using diazo reagent (Ehrlich's diazo reagent).
- Conjugated (direct) bilirubin is water-soluble and reacts immediately and directly with the diazo reagent → DIRECT positive.
- Unconjugated (indirect) bilirubin is lipid-soluble and bound to albumin; it requires alcohol to first dissolve it before reacting → gives only indirect positive.
- In obstructive jaundice: Bile duct obstruction → conjugated bilirubin (already processed by liver) cannot be excreted → dams back into hepatic sinusoids → regurgitates into blood → elevated serum conjugated bilirubin → Van den Bergh DIRECT positive.
- Serum bilirubin is 6 mg/dl (normal <1 mg/dl), predominantly direct (conjugated).
CASE 12 (Page 12) - CIRCULATORY SHOCK (HYPOVOLEMIC)
Case Summary
Accident victim | Restlessness, extreme weakness | Pale, cold, clammy skin | Rapid thready pulse | Hypotension | Oliguria
Symptom Analysis & Differential Diagnosis Reasoning
This is a trauma victim with the complete picture of hypovolemic (hemorrhagic) shock. Shock is defined as inadequate tissue perfusion to meet metabolic demands.
Pathophysiology cascade:
Trauma → Blood loss → ↓ Blood volume → ↓ Venous return → ↓ Cardiac output → ↓ BP → Baroreceptors activated → Sympathetic surge + RAAS activation → Compensatory responses (but if severe → decompensation)
Symptoms explained:
| Symptom | Mechanism |
|---|
| Restlessness | Cerebral hypoperfusion → neuronal irritability; anxiety from pain and hypoxia |
| Extreme weakness | Reduced cardiac output → reduced muscle perfusion → energy deficit |
| Pale skin | Sympathetic vasoconstriction of cutaneous vessels → diversion of blood to vital organs (heart, brain) → skin pallor |
| Cold skin | Cutaneous vasoconstriction → reduced skin blood flow → skin temperature drops |
| Clammy skin | Sympathetic activation of eccrine sweat glands (adrenergic) → profuse sweating |
| Rapid thready pulse | Rapid: Compensatory tachycardia (sympathetic ↑ HR). Thready: Low pulse volume/pressure due to low cardiac output + intense vasoconstriction |
| Hypotension | Reduced circulating blood volume → reduced preload → reduced cardiac output → BP falls; if <90 mmHg systolic = clinical shock |
| Oliguria | ↓ Renal perfusion pressure → ↓ GFR → ↓ urine output; also RAAS → ADH → water retention; a protective mechanism but indicates severe hypoperfusion |
Answers
Q1. Provisional diagnosis:
Hypovolemic (Hemorrhagic) Shock - Class III-IV.
Q2. Explanation of symptoms and signs:
(See pathophysiology table above - expanded below)
Cold, pale, clammy skin: When blood pressure falls, baroreceptors in the carotid sinus and aortic arch detect the fall and signal the vasomotor center in the medulla. The sympathetic nervous system is activated massively. This causes:
- Vasoconstriction of skin and splanchnic vessels (α1 receptors) → skin becomes pale and cold
- Activation of sweat glands (cholinergic sympathetic fibers) → clammy skin
Rapid thready pulse: Sympathetic stimulation increases heart rate (β1 receptors on SA node → tachycardia). The pulse is "thready" (weak, barely palpable) because the pulse pressure (systolic - diastolic BP) is very narrow due to low stroke volume (↓ preload from blood loss) and intense vasoconstriction (elevated diastolic BP).
Hypotension: Blood volume loss → reduced venous return → reduced end-diastolic volume (preload) → reduced stroke volume (Frank-Starling mechanism) → reduced cardiac output → reduced MAP (MAP = CO × TPR). Despite increased TPR (vasoconstriction), cardiac output is so low that BP falls.
Oliguria: Renal blood flow falls in proportion to cardiac output. ↓ Renal perfusion → ↓ GFR → less filtrate → less urine. Simultaneously: ↓ BP → macula densa detects → JGA releases renin → Angiotensin II → Aldosterone → Na⁺ and water retention. Also, hypovolemia + hyperosmolality → ADH release from posterior pituitary → water reabsorption in collecting duct. Combined effect: severe oliguria.
Restlessness: Brain has no significant glycogen stores and no vasoconstriction (cerebral vasculature auto-regulates, but in severe shock, CBF falls) → neuronal hypoxia → agitation, confusion, restlessness.
Q3. Immediate treatment:
- Arrest the bleeding (if external): Direct pressure, tourniquet if limb hemorrhage.
- Airway, Breathing, Circulation (ABC): Secure airway, administer 100% O₂.
- IV access: Two large-bore (14-16G) peripheral IV cannulas immediately.
- IV fluid resuscitation:
- Initial bolus: Crystalloids - Normal saline or Ringer's Lactate - 1-2 liters rapidly
- For hemorrhagic shock: Blood transfusion - Packed RBCs (type-matched or O-negative emergency)
- Colloids (albumin, hetastarch) for plasma volume expansion
- Position: Trendelenburg position (legs elevated 15-20°) to improve venous return.
- Monitor: Heart rate, BP, urine output (Foley catheter), CVP, oxygen saturation continuously.
- Vasopressors (only after adequate volume replacement): Noradrenaline (norepinephrine) or dopamine if hypotension persists.
- Treat underlying cause: Emergency surgery if internal hemorrhage.
CASE 13 (Page 13) - RIGHT-SIDED HEART FAILURE (COR PULMONALE / RHF)
Case Summary
Patient | Leg swelling worse in evening | Dependent pitting edema | Distended neck veins | Enlarged liver (soft, tender - hepatomegaly) | Oliguria and nocturia | Raised JVP | Urine albumin: present | X-ray: Right ventricular hypertrophy | ECG: Elevated P wave
Symptom Analysis & Differential Diagnosis Reasoning
This is a textbook presentation of Right-Sided Heart Failure (RHF). The right ventricle fails to pump blood forward into the pulmonary circulation → blood backs up into the systemic veins → increased systemic venous pressure → venous congestion.
How to rule out Left Heart Failure:
- Left heart failure: primarily causes pulmonary congestion → dyspnea, orthopnea, pulmonary edema, pink frothy sputum, cardiomegaly (left side), S3 gallop. JVP not elevated (right side is not backing up).
- This patient has no pulmonary symptoms but has systemic venous congestion → Right heart failure.
How to rule out liver cirrhosis (also causes edema + hepatomegaly):
- Cirrhosis: ascites prominent, spider angiomas, caput medusae, splenomegaly, low albumin, elevated liver enzymes. JVP is NOT raised. X-ray normal.
- This patient: JVP raised, RV hypertrophy on X-ray → cardiac cause confirmed.
How to rule out nephrotic syndrome (also edema + proteinuria):
- Nephrotic: massive proteinuria (>3.5g/day), hypoalbuminemia, hypercholesterolemia. JVP normal. No hepatomegaly. No RV hypertrophy.
- This patient has only trace albumin in urine (secondary to congestion) and RV hypertrophy.
Answers
Q1. Diagnosis:
Right-Sided Congestive Heart Failure (RHF) - likely Cor Pulmonale (RHF secondary to pulmonary hypertension/lung disease) given the RV hypertrophy.
Key diagnostic features:
- Dependent pitting edema (worse in evening - gravitational)
- Raised JVP (systemic venous hypertension)
- Hepatomegaly (congestive hepatopathy - "nutmeg liver")
- Oliguria + nocturia
- X-ray: RV hypertrophy
- ECG: Elevated P wave (P pulmonale)
Q2. How do you explain dependent edema and liver enlargement?
Dependent Pitting Edema:
- Right heart failure → right ventricle cannot pump all blood forward → blood accumulates in the right atrium and systemic veins
- Systemic venous pressure rises → increased hydrostatic pressure in peripheral capillaries (especially in legs/ankles due to gravity)
- Starling forces: Normally, hydrostatic pressure pushes fluid out of capillaries, and oncotic pressure pulls it in. When hydrostatic pressure rises chronically, net filtration exceeds lymphatic drainage → fluid accumulates in interstitial spaces → pitting edema.
- It worsens toward evening because the patient has been upright all day (gravity accumulates fluid in dependent areas). It improves in the morning after lying down overnight (fluid redistributes).
- Simultaneously, secondary hyperaldosteronism (RAAS activation due to low cardiac output) causes Na⁺ and water retention → further worsens edema.
- Low albumin (from hepatic congestion) reduces oncotic pressure → less fluid pull-back → exacerbates edema.
Hepatomegaly (Congestive Hepatopathy):
- The liver is drained by the hepatic veins which drain into the inferior vena cava (IVC) → right atrium.
- In RHF, right atrial pressure rises → IVC pressure rises → hepatic venous pressure rises → blood cannot drain from the liver sinusoids → sinusoidal congestion.
- Liver becomes engorged with blood → enlarged, soft, and tender (the stretching of Glisson's capsule causes tenderness).
- Chronic congestion leads to hepatocyte hypoxia (especially centrilobular - zone 3 furthest from portal blood supply) → nutmeg liver pattern on histology → eventually cardiac cirrhosis if untreated.
Q3. Why is the P wave elevated in ECG?
The P wave represents atrial depolarization (both atria).
- In Right Heart Failure (especially Cor Pulmonale), there is pulmonary hypertension → right ventricle works against high resistance → right ventricle hypertrophies → pressure is transmitted back to the right atrium → right atrial hypertrophy/enlargement → the right atrium must generate a stronger depolarization wave to empty against elevated RV pressure.
- This produces P pulmonale: Tall, peaked P wave (>2.5 mm in lead II) in ECG.
- Normally the P wave is <2.5 mm tall. Right atrial enlargement produces taller, narrower (peaked) P waves, especially visible in leads II, III, and aVF.
- This is distinct from P mitrale (bifid, broad P wave in left atrial enlargement, seen in mitral stenosis).
CASE 14 (Page 14) - ANGINA PECTORIS (STABLE)
Case Summary
Patient | Severe constricting pain behind the sternum | Pain radiates along ulnar border of left upper limb | Severity proportional to exertion | X-ray chest: Normal | ECG at rest: Normal (nothing suggestive)
Symptom Analysis & Differential Diagnosis Reasoning
Pain characteristics - classic Angina Pectoris:
- Location: Retrosternal (behind sternum) - the heart is in the mediastinum
- Character: Constricting/squeezing - from ischemic myocardium
- Radiation: Along the inner (ulnar) border of the left arm - via T1-T4 dermatomes (same spinal segments as cardiac sympathetic afferents) - referred pain
- Trigger: Exertion - increased myocardial O₂ demand exceeds supply through narrowed coronary arteries
- Relief: Rest (reduces demand), or nitrates (vasodilate coronary vessels)
How to rule out other causes:
| Condition | Key differentiating features |
|---|
| Myocardial Infarction (MI) | Pain at rest, prolonged (>30 min), NOT relieved by rest alone, ECG changes (ST elevation), troponin elevated |
| GERD/esophageal spasm | Burning, related to food/lying down, relieved by antacids |
| Costochondritis | Localized tenderness on pressing the costochondral junction |
| Pulmonary embolism | Pleuritic pain (worse with breathing), dyspnea, tachycardia, hypoxia |
| Aortic dissection | Sudden tearing pain radiating to back, BP difference between arms |
| Pericarditis | Positional (better sitting forward), friction rub on auscultation |
Here: Exertional, constricting, retrosternal, radiating to left arm, relieved by rest + Normal X-ray + Normal resting ECG = Classic Stable Angina Pectoris (from coronary artery disease / atherosclerosis).
Answers
Q1. Diagnosis:
Stable Angina Pectoris (due to coronary artery disease / atherosclerosis of coronary vessels).
The clinical features are pathognomonic:
- Retrosternal constricting chest pain
- Radiation to left arm (ulnar border) - referred pain via T1-T4 dermatomes
- Proportional to exertion (demand ischemia)
- (Implied) relief with rest
Q2. Why was ECG normal?
The ECG is taken at rest, and there are very important reasons it is normal in stable angina:
- Angina occurs during exertion, not at rest. The coronary arteries are narrowed (stenosed) by atherosclerotic plaque, but not completely occluded. At rest, blood flow through the narrowed vessel is still adequate to meet the low myocardial oxygen demand → no ischemia → no ECG changes.
- ECG changes in angina occur only during ischemia (exercise/stress): During ischemia, ventricular repolarization is altered → ST depression (or elevation in variant/Prinzmetal angina), T-wave inversion appear. These changes are transient and revert with cessation of ischemia.
- In MI (complete occlusion, permanent ischemia), ECG changes are permanent: ST elevation (STEMI), Q waves, T-wave inversion.
- Chest X-ray is normal: Stable angina does not cause heart enlargement (unlike chronic heart failure). The coronary vessels are not visible on plain chest X-ray. There is no pulmonary congestion at rest.
- To "unmask" the ECG changes in angina: Stress ECG (Exercise Tolerance Test / TMT - Treadmill Test) is performed. When the patient exercises to 85% of maximum heart rate, demand exceeds supply → ischemia → >1 mm ST depression in ECG = positive test = confirms coronary artery disease.
Q3. Advice to such a patient:
Immediate management:
- Sublingual nitroglycerine (GTN): To be taken immediately when pain occurs - dilates coronary arteries and systemic veins (reduces preload and afterload → reduces myocardial oxygen demand). Carry at all times.
Lifestyle modification:
- Stop smoking - smoking accelerates atherosclerosis and causes coronary vasospasm.
- Dietary changes: Low saturated fat, low cholesterol diet to slow atherosclerosis progression.
- Regular moderate exercise: Paradoxically, regular graded exercise training increases collateral coronary circulation over time and improves exercise tolerance.
- Weight reduction if overweight - reduces cardiac work.
- Control blood pressure - hypertension accelerates coronary disease.
- Stress management: Emotional stress can trigger angina (catecholamine surge increases HR and BP → demand ↑).
Medical treatment:
- Beta-blockers (atenolol, metoprolol): Reduce heart rate and contractility → reduce myocardial O₂ demand → prevent angina.
- Long-acting nitrates (isosorbide mononitrate): Prophylaxis.
- Calcium channel blockers (amlodipine): Coronary vasodilation.
- Aspirin (75-150 mg/day): Antiplatelet - prevents thrombus formation on plaques.
- Statins (atorvastatin): Reduce LDL cholesterol and stabilize atherosclerotic plaques.
Investigation:
- Treadmill test / Stress ECG: To assess severity and document ischemia.
- Coronary angiography: To visualize the extent and location of stenosis - guides decision for angioplasty (PTCA) or bypass surgery (CABG).
Sources: Guyton and Hall Textbook of Medical Physiology | Ganong's Review of Medical Physiology | Robbins & Kumar Basic Pathology | Costanzo Physiology 7th Edition | Medical Physiology (Boron & Boulpaep) | Basic Medical Biochemistry (Harvey)