Hsts ammonia
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive information about ammonia from multiple authoritative textbooks. Let me compose a thorough answer. Note that "Hsts" is most likely a typo or autocorrect for "HSTs" - possibly meaning "tests" (i.e., the user is asking about ammonia tests/lab values), or it could be "hosts" or simply a garbled prefix. Given the lab-focused results and context, I'll provide a complete clinical overview of ammonia covering biochemistry, metabolism, lab measurement, and clinical significance.
Ammonia - Clinical Overview
What Is Ammonia?
Ammonia (NH3/NH4+) is a nitrogenous compound derived primarily from amino acid and nucleic acid catabolism. It is toxic - particularly to the brain - and must be rapidly detoxified. At physiological pH (~7.4), the vast majority (~98.3%) exists as the ionized form NH4+, with only ~1.7% as the free NH3 form. Small changes in pH cause exponential shifts in the NH3:NH4+ ratio, which has important implications for transport and toxicity.
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 528
Urea Cycle (Krebs-Henseleit Cycle)
Ammonia is detoxified exclusively in the liver via the urea cycle. The liver uniquely possesses all the required enzymes:
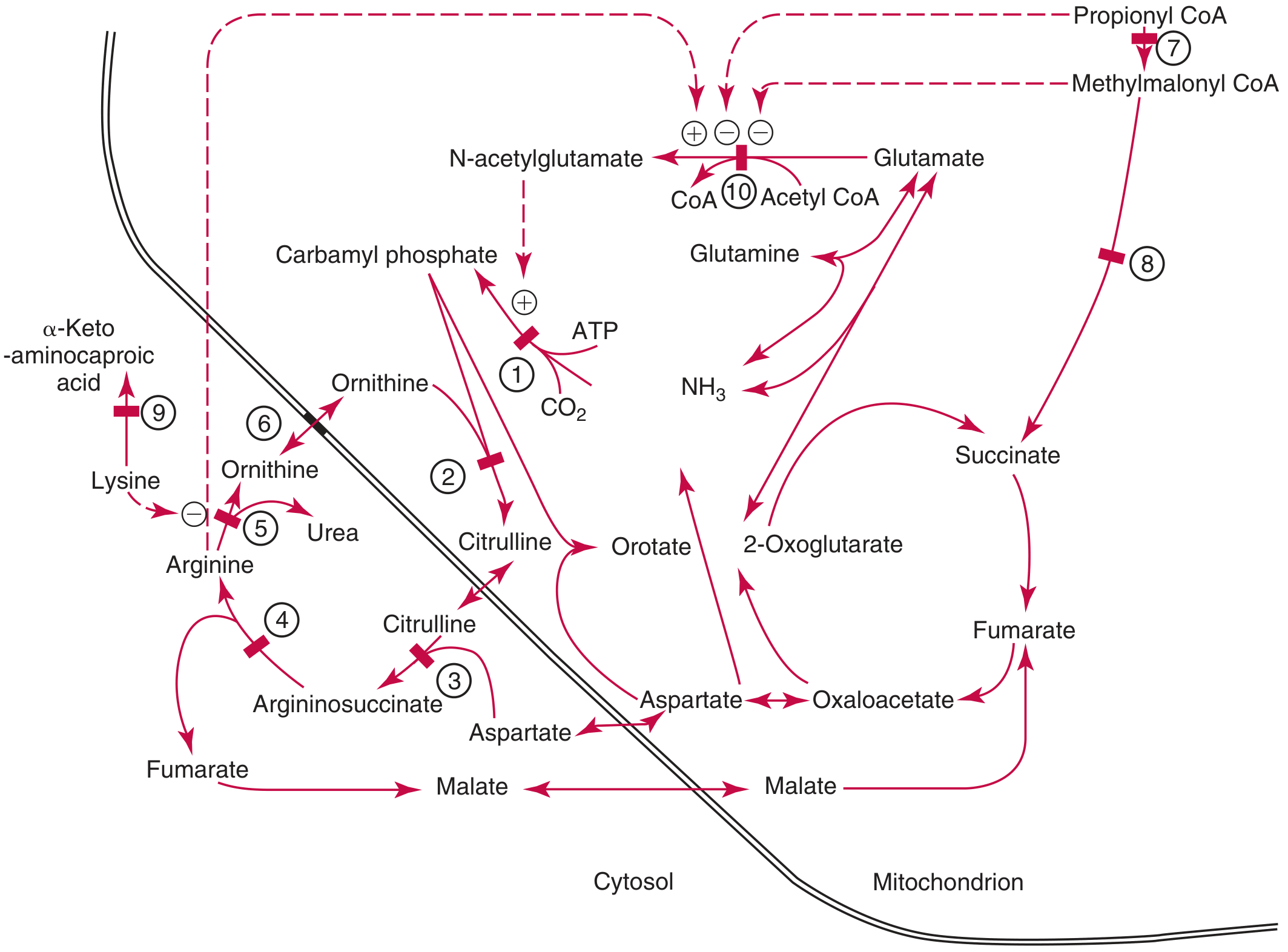
Key steps (mitochondria -> cytosol):
- NH3 + CO2 + ATP -> Carbamoyl phosphate (enzyme: CPS-1 - rate-limiting step)
- Carbamoyl phosphate + Ornithine -> Citrulline (enzyme: OCT - unique to liver)
- Citrulline + Aspartate -> Argininosuccinate (enzyme: ASS)
- Argininosuccinate -> Arginine + Fumarate (enzyme: ASL)
- Arginine -> Ornithine + Urea (enzyme: Arginase) - Urea is then renally excreted
Congenital deficiency of any of these enzymes causes hyperammonemia (urea cycle disorders). The numbered defect sites visible in the diagram correspond to: (1) CPS-1, (2) OCT, (3) ASS, (4) ASL, (5) Arginase, (6) mitochondrial ornithine transport, (7) propionyl-CoA carboxylase, (8) methylmalonyl-CoA mutase, (9) L-lysine dehydrogenase, and (10) N-acetylglutamate synthetase.
- Tietz Textbook of Laboratory Medicine, 7th Edition, p. 1977-1978
Renal Ammonia Metabolism
The kidney is a net producer of ammonia (renal vein ammonia > arterial ammonia). This is critical for acid-base homeostasis:
-
Primary site: Proximal tubule (60-70% of renal ammoniagenesis under basal conditions; 70-80% during metabolic acidosis)
-
Key enzyme: Phosphate-dependent glutaminase (PDG/KGA), which converts glutamine -> glutamate + NH4+
-
Further processing: Glutamate dehydrogenase (GDH) converts glutamate -> alpha-ketoglutarate + NH4+
-
NH3 chemistry: NH4+ can substitute for K+ at K+-transport sites due to similar biophysical properties in aqueous solution
-
Ammonia is selectively transported to the urine (for acid excretion) or to the renal vein; integrated transport occurs in the proximal tubule, thick ascending limb (TAL), and collecting duct
-
Brenner and Rector's The Kidney, 2-Volume Set
Causes of Hyperammonemia
| Category | Examples |
|---|---|
| Liver disease | Cirrhosis, fulminant hepatic failure, Reye syndrome |
| Portosystemic shunting | Intrahepatic (cirrhosis) or surgical shunts |
| Urea cycle enzyme defects | CPS-1 deficiency, OTC deficiency, citrullinemia, etc. |
| Organic acidemias | Propionic acidemia, methylmalonic acidemia |
| Drugs | Valproate, salicylates |
| Renal failure | Increased urea diffuses into GI tract -> bacterial conversion to NH3 |
| GI bleeding | Bacterial metabolism of protein in blood |
| Excess dietary protein / constipation | Increased gut ammonia production |
- Tietz Textbook of Laboratory Medicine, 7th Edition, p. 1978
Ammonia Toxicity: Mechanisms in the CNS
Ammonia causes hepatic encephalopathy through several mechanisms:
- GABA depletion: Ammonia reacts with glutamic acid to form glutamine (reversing glutaminase). This depletes glutamate, which is the precursor for GABA synthesis, leading to reduced inhibitory neurotransmission.
- Astrocyte toxicity: Ammonia is directly toxic to astrocytes.
- Glutamine accumulation: Acts as an osmolyte contributing to cerebral edema.
- Synergy with inflammation and oxidative stress: Complex interaction that amplifies neurotoxicity.
Key clinical note: Arterial ammonia correlates better with encephalopathy severity than venous levels. Lowering ammonia invariably reduces encephalopathy. In acute liver failure, ammonia >200 µmol/L (340 µg/dL) is associated with cerebral edema and poor prognosis.
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 528-529
Laboratory Measurement
Method: Enzymatic assay using glutamate dehydrogenase (GLDH):
- NH3 + alpha-ketoglutarate + NADPH -> Glutamate + NADP+
- Ammonia concentration is determined by the decrease in absorbance at 340 nm (as NADPH -> NADP+)
Reference interval (enzymatic method):
- 15 to 45 µg/dL (11 to 32 µmol/L)
Key interferences and pre-analytical issues:
- Hemolysis - RBCs contain ~3x the ammonia of plasma; falsely elevates results
- Cigarette smoke - contains ammonia; keep samples away from smoke
- ALT up to 2400 U/L does not interfere
- Sample stability: 3 hours at 2-4°C, or 24 hours at -20°C
- Fasting venous plasma sample is preferred
Clinical utility:
-
Useful in differential diagnosis of encephalopathy (to determine if hepatic origin)
-
Especially helpful in diagnosing Reye syndrome and inherited urea cycle disorders
-
NOT useful for monitoring encephalopathy severity in known chronic liver disease (poor correlation at that stage; ~70% of cirrhotic patients without encephalopathy already have elevated levels)
-
Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 529; Tietz Textbook of Laboratory Medicine, 7th Edition, p. 1978-1979
This is a shared conversation. Sign in to Orris to start your own chat.