Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching Images
acute respiratory distress syndrome pathogenesis mechanism diagram
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Searching the Web
ARDS pathogenesis mechanism neutrophil alveolar damage phases 2024
Loading Image
Reading Web Page
https://www.ncbi.nlm.nih.gov/books/NBK436002/
I now have comprehensive information to provide a thorough answer. Let me synthesize everything:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition and Diagnostic Criteria (Berlin 2012)
ARDS is an acute, diffuse inflammatory lung injury defined by:
- Bilateral pulmonary infiltrates on CXR/CT not explained by effusions, collapse, or nodules
- PaO₂/FiO₂ ratio < 300 mmHg (mild: 200–300; moderate: 100–200; severe: <100) on PEEP ≥ 5 cm H₂O
- Onset within 7 days of a known clinical insult
- Not fully explained by cardiac failure or fluid overload
Common precipitants — direct (pneumonia, aspiration, inhalation injury) and indirect (sepsis, severe pancreatitis, major trauma, massive transfusion, burns).
Core Pathological Process: Diffuse Alveolar Damage (DAD)
ARDS progresses through three overlapping histological phases:
Phase 1 — Exudative (Days 0–7)
Initiating event: A direct lung insult or systemic inflammatory signal (e.g., endotoxin in sepsis, pancreatic enzymes in pancreatitis) activates the innate immune response.
Key cellular events:
-
Macrophage activation — Alveolar macrophages recognize PAMPs/DAMPs via Toll-like receptors (TLR) and pattern recognition receptors (PRR), switching to the M1 (pro-inflammatory) phenotype. They release:
- TNF-α, IL-1β, IL-6, IL-8 (CXCL8)
- Reactive oxygen species (ROS)
- Platelet-activating factor
-
Neutrophil recruitment and sequestration — IL-8 and complement fragments (C5a) drive massive neutrophil recruitment into alveolar capillaries. Activated neutrophils undergo cytoskeletal rearrangements, becoming less deformable and physically sequestering in the narrow alveolar capillaries. Once adherent (via ICAM-1/β₂-integrin interactions), they transmigrate into the interstitium and alveolar space.
-
Neutrophil effector mechanisms — Extravasated neutrophils cause epithelial and endothelial injury through:
- Proteases (elastase, matrix metalloproteinases) — degrade basement membrane
- ROS / oxidants — oxidative stress to cell membranes
- Myeloperoxidase (MPO) — generates hypochlorous acid
- Neutrophil extracellular traps (NETs) — trap microbes but also damage endothelium and activate platelets
-
Platelet–neutrophil complexes — Activated platelets bind neutrophils via P-selectin/PSGL-1, amplifying ROS and NET release, further injuring the alveolocapillary membrane.
-
T-cell imbalance — Pro-inflammatory macrophages shift T-cell responses toward Th17 (↑ IL-17) and away from T-regulatory cells (↓ IL-10, ↓ TGF-β), sustaining the inflammatory milieu and suppressing resolution.
Structural consequences:
| Structure | Damage | Result |
|---|---|---|
| Capillary endothelium | Injury → gaps in tight junctions | Protein-rich fluid leaks into interstitium |
| Type I alveolar epithelium | Necrosis/apoptosis (necroptosis, pyroptosis) | Loss of air–blood barrier integrity |
| Type II alveolar epithelium | Dysfunction/injury | ↓ Surfactant production → alveolar collapse |
| Alveolar space | Flooding with edema fluid, fibrin, RBCs | Impaired gas exchange, hyaline membrane formation |
Hyaline membranes — the histological hallmark of DAD — form from proteinaceous exudate, fibrin, and cellular debris lining the alveolar walls, compounding gas diffusion failure.
Physiological result: Non-cardiogenic pulmonary edema → ↓ FRC → intrapulmonary shunt → refractory hypoxemia. Lung compliance falls ("stiff lungs") due to edema and surfactant loss.
Phase 2 — Proliferative (Days 3–14)
- Type II pneumocyte hyperplasia — surviving type II cells proliferate to repopulate denuded alveolar walls and restore surfactant production
- Fibroblast activation — myofibroblasts migrate into alveolar spaces and interstitium; fibronectin and TGF-β drive collagen deposition
- Inflammation may partially resolve with macrophage phenotype shift toward M2 (anti-inflammatory, pro-repair)
- Lung function may begin to improve; some patients wean from ventilation
Phase 3 — Fibrotic (Day 14+)
- Extensive intra-alveolar fibrosis replaces the hyaline membrane exudate
- Persistent architectural distortion → chronic restrictive physiology, impaired gas exchange
- Occurs in severe cases; associated with prolonged ventilator dependence and worse long-term outcomes
- Patients who survive may have permanent exercise intolerance and reduced quality of life
Immunothrombosis
An important, emerging mechanism: the coagulation and innate immune systems interact extensively in ARDS. Complement activation (C5a/C5aR1) amplifies platelet-mediated thrombogenicity in a NET-dependent manner, causing pulmonary microthrombosis. This further reduces perfusion and worsens ventilation–perfusion mismatch.
Gas Exchange Failure: Physiological Summary
Alveolar flooding + surfactant loss
↓
Alveolar collapse (atelectasis) + low compliance
↓
Intrapulmonary shunt (perfusion without ventilation)
↓
Refractory hypoxemia (PaO₂/FiO₂ < 300)
↓
Compensatory hyperventilation → hypocapnia (early)
↓
Respiratory muscle fatigue → hypercapnic respiratory failure (late)
The injury is non-uniform — dependent zones (lung bases) are worst affected due to gravitational edema, while non-dependent zones may be relatively normal. This heterogeneity ("baby lung") is why high PEEP can recruit collapsed alveoli in some regions while causing overdistension and volutrauma in adjacent, healthier ones.
Pulmonary Vascular Effects
- Hypoxic vasoconstriction + vascular remodeling → pulmonary hypertension
- Increased right ventricular afterload → possible RV strain/cor pulmonale
- Microvascular thrombosis further raises pulmonary vascular resistance
Pathogenesis Diagram
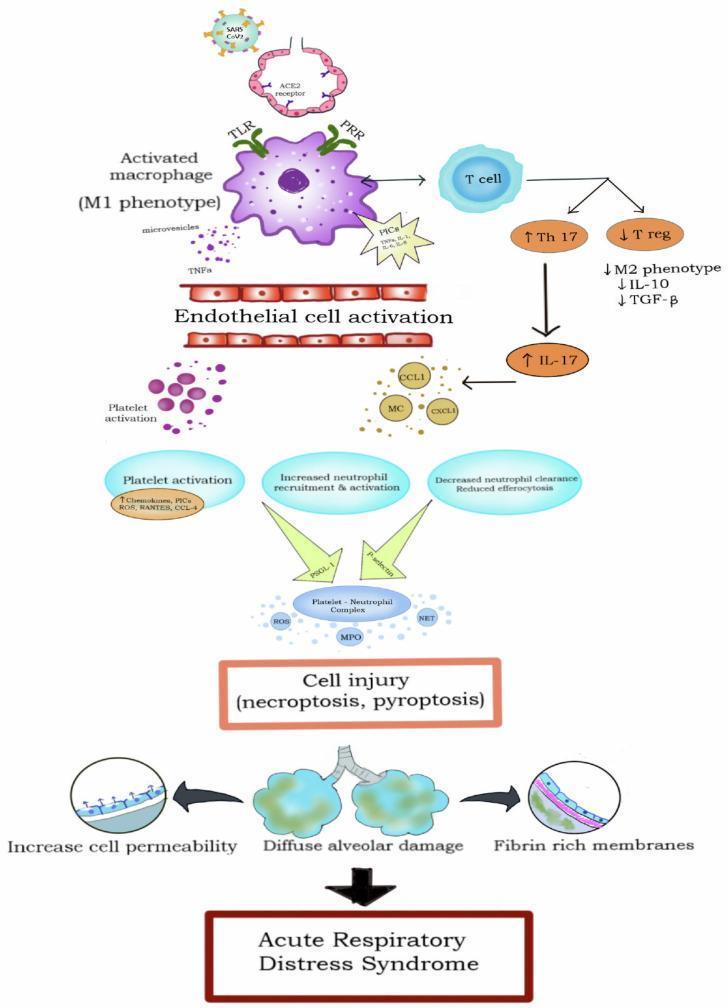
Macrophage activation (M1) → cytokine storm → endothelial activation → platelet–neutrophil complexes → ROS/MPO/NETs → cell injury (necroptosis/pyroptosis) → diffuse alveolar damage, increased permeability, fibrin-rich hyaline membranes → ARDS
Key Mediators Summary
| Mediator | Source | Role in ARDS |
|---|---|---|
| TNF-α, IL-1β, IL-6 | Macrophages | Systemic and local inflammation, endothelial activation |
| IL-8 (CXCL8) | Macrophages, epithelium | Primary neutrophil chemoattractant |
| IL-17 | Th17 cells | Sustains neutrophilic inflammation |
| Elastase, MPO | Neutrophils | Direct tissue proteolysis and oxidant injury |
| NETs | Neutrophils | Endothelial damage, platelet activation, microthrombosis |
| Phospholipase A₂ | Pancreatic (in pancreatitis) | Surfactant degradation |
| ROS | Neutrophils, macrophages | Membrane lipid peroxidation |
| TGF-β | Multiple | Fibroblast activation → fibrosis |
Sources: Murray & Nadel's Textbook of Respiratory Medicine; Goldman-Cecil Medicine; Sabiston Textbook of Surgery; StatPearls (ARDS); Nature Signal Transduction 2025 review on ARDS pathophysiology and heterogeneity.
This is a shared conversation. Sign in to Orris to start your own chat.