Feline esophagitis History of food allergy IL-5 Diagnosis requires more than 15 eosinophils per high power field
Reading File
Finding Sources
Finding Sources
Searching PubMed
"eosinophilic esophagitis"[MeSH Terms] AND diagnosis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
All the key information is in hand. Here is the complete answer:
Eosinophilic Esophagitis (EoE)
The four clues you listed - feline/ringed esophagus appearance, history of food allergy, IL-5 involvement, and the >15 eosinophils/HPF threshold - all point to Eosinophilic Esophagitis (EoE).
Definition
EoE is a chronic immune-mediated inflammatory disorder characterized by:
-
Esophageal dysfunction symptoms (dysphagia, food impaction in adults; feeding intolerance, GERD-like symptoms in children)
-
Histological eosinophilic infiltration of the esophageal epithelium
-
Yamada's Textbook of Gastroenterology, 7e
-
Robbins & Kumar Basic Pathology
Pathophysiology & Role of IL-5
EoE arises from an exaggerated Th2-mediated allergic immune response to food (and aeroallergens). Key cytokines involved:
| Cytokine | Role |
|---|---|
| IL-5 | Major eosinophil growth factor, survival signal, and recruiter - drives eosinophilic infiltration of the esophagus |
| IL-13 | Induces esophageal epithelial barrier disruption and eotaxin-3 production |
| IL-33 / TSLP | Activates T-regulatory and Th2 cells, initiating the inflammatory cascade |
| Eotaxin-3 | Chemokine that directly recruits eosinophils to the esophagus |
IL-5 is so central that anti-IL-5 biologics (mepolizumab, reslizumab) have been studied as targeted therapies. - Yamada's Textbook of Gastroenterology, 7e, IL-4/IL-5/IL-13 section
Link to Food Allergy
-
Most patients have a personal or family history of atopic disease: allergic rhinitis, asthma, atopic dermatitis, or food allergy
-
Food antigen sensitization (particularly cow's milk, wheat, soy, eggs, nuts, seafood) is a major trigger
-
Sensitization often occurs via inflamed skin (the "outside-in" hypothesis), with re-exposure through ingestion driving esophageal inflammation
-
Removal of trigger foods via elemental or elimination diets results in histological remission in a significant proportion of patients
-
Sleisenger & Fordtran's GI and Liver Disease
Diagnostic Criteria
Diagnosis requires BOTH clinical and histological criteria:
- Symptoms of esophageal dysfunction
- Histology: ≥15 eosinophils per high-power field (eos/HPF) on esophageal biopsy (proximal AND distal biopsies)
- Exclusion of other causes of esophageal eosinophilia (GERD, parasitic infection, Crohn disease, drug-induced esophagitis, eosinophilic gastroenteritis)
- PPI-refractory course (helps distinguish from GERD-related eosinophilia)
"The eosinophil density required for the diagnosis of eosinophilic esophagitis is generally more than 15 eosinophils per high-power field on both proximal and distal esophageal biopsies." - Cummings Otolaryngology, Head & Neck Surgery
"The presence of at least 15 eosinophils per HPF is required for a diagnosis, and it is also a cut-off for histological remission (<15 eos/HPF)." - Yamada's Textbook of Gastroenterology, 7e
The eosinophilic infiltrate is often patchy, so multiple biopsies (at least 2-4 from proximal and distal esophagus) are recommended to avoid false negatives (~10% of biopsies nondiagnostic due to patchiness).
Endoscopic & Histologic Findings
Here are the classic findings side by side:
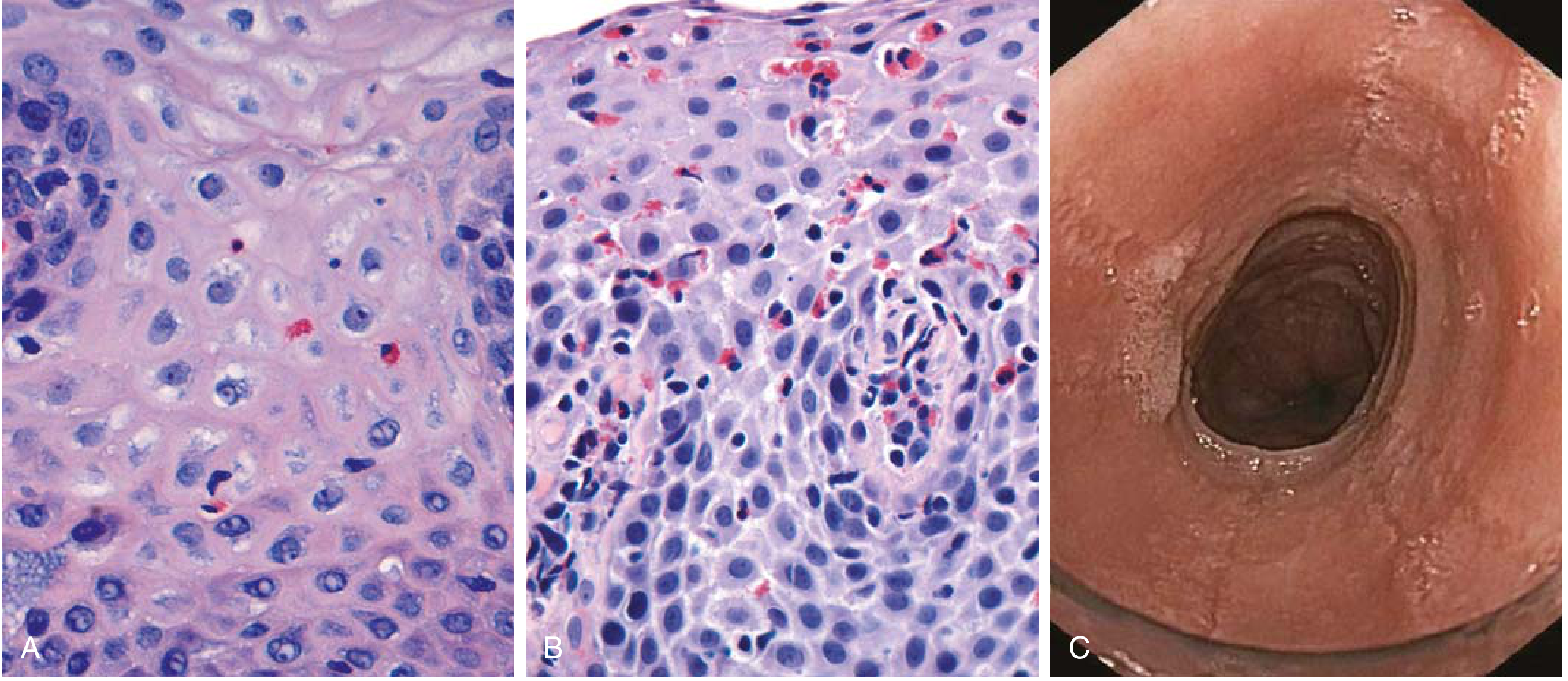
Fig. 13.9 - Robbins & Kumar Basic Pathology: (A) Reflux esophagitis - scattered eosinophils; (B) EoE - numerous eosinophils with eosinophilic microabscesses (superficial clustering); (C) Endoscopy showing circumferential rings ("feline esophagus" / trachealization)
Endoscopic features:
- Stacked concentric rings ("trachealization" or "feline esophagus") - fixed or transient
- Longitudinal furrows
- White exudates / eosinophilic microabscesses (can mimic candidiasis)
- Mucosal friability ("crepe paper esophagus")
- Strictures (proximal/mid esophagus - distinguishes from GERD which affects distal)
Histologic features:
- ≥15 eos/HPF (superficial clustering is characteristic)
- Eosinophilic microabscesses
- Basal cell hyperplasia
- Lamina propria fibrosis (subepithelial)
Key Differentiator from GERD
| Feature | EoE | GERD |
|---|---|---|
| Location | Proximal/mid esophagus | Distal esophagus |
| Eosinophils | ≥15/HPF, superficial clusters | Scattered, usually <10/HPF |
| PPI response | Refractory | Responsive |
| Atopic history | Common | Not typical |
| Rings/furrows | Present | Absent |
Treatment
- Dietary elimination (6-food elimination diet or elemental formula)
- Swallowed topical corticosteroids (fluticasone, budesonide) - first-line pharmacotherapy
- Proton pump inhibitors - can induce remission in a subset (~30-40%, per recent meta-analysis, PMID 40089255)
- Dupilumab (anti-IL-4Rα, blocks IL-4 and IL-13 signaling) - FDA-approved biologic; real-world effectiveness confirmed in systematic review (PMID 39998943)
- Esophageal dilation for strictures/food impaction
Protein losing enteropathy Cerebriform appearance Menetrier ds Case study
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
All the content needed is in hand. Here is the complete structured answer:
Ménétrier's Disease
The clues - protein-losing enteropathy + cerebriform (brain-like) appearance + gastric pathology - are the classic triad of Ménétrier's Disease, a rare hypertrophic gastropathy.
Definition
Ménétrier's disease (MD) is a rare disorder of the stomach characterized by:
-
Massive foveolar (mucous cell) hyperplasia of the body and fundus
-
Giant, tortuous gastric rugal folds with a cerebriform (brain-like) gross appearance
-
Protein-losing gastropathy with hypoalbuminemia
-
Achlorhydria / hypochlorhydria due to loss of parietal cell mass
-
Robbins, Cotran & Kumar Pathologic Basis of Disease
-
Harrison's Principles of Internal Medicine, 22e
Pathogenesis
The molecular driver is overexpression of TGF-α (Transforming Growth Factor-alpha):
TGF-α overexpression
↓
Overstimulation of EGFR (Epidermal Growth Factor Receptor) on gastric epithelial cells
↓
Massive proliferation of foveolar (surface mucous) cells
↓
Hyperplastic, elongated, tortuous, cystic glands → Giant rugal folds
↓
Loss of parietal cells → Hypochlorhydria/Achlorhydria
Loss of protein across leaky mucosa → Hypoalbuminemia → Edema
-
In children: most cases are triggered by CMV infection (CMV-induced EGFR activation) - typically self-limited
-
In adults: etiology often unknown; CMV and H. pylori have also been implicated
-
Yamada's Textbook of Gastroenterology, 7e
-
Harrison's Principles of Internal Medicine, 22e
Clinical Presentation
Demographics: Typically men, ages 40-60 years (male predominance)
Symptoms (insidious and progressive):
- Epigastric pain, nausea, vomiting, anorexia
- Peripheral edema - from hypoalbuminemia (albumin loss can approach 10 g/day)
- Weight loss, diarrhea
- Occult GI bleeding (from mucosal erosions)
20-100% of patients develop protein-losing gastropathy with hypoalbuminemia (depending on stage at presentation).
Classic Case Scenario
A 55-year-old man presents with progressive peripheral edema, epigastric discomfort, and weight loss over several months. Labs show hypoalbuminemia with a normal 24-hour urine protein. Upper endoscopy reveals massively enlarged, lobular, cerebriform gastric folds in the fundus and body with antral sparing. Biopsy shows massive foveolar hyperplasia with elongated, corkscrew-shaped, cystic glands and markedly reduced parietal and chief cells. Gastric acid secretion is absent.Diagnosis: Ménétrier's Disease
Radiology
The barium meal/upper GI study is dramatic and characteristic:
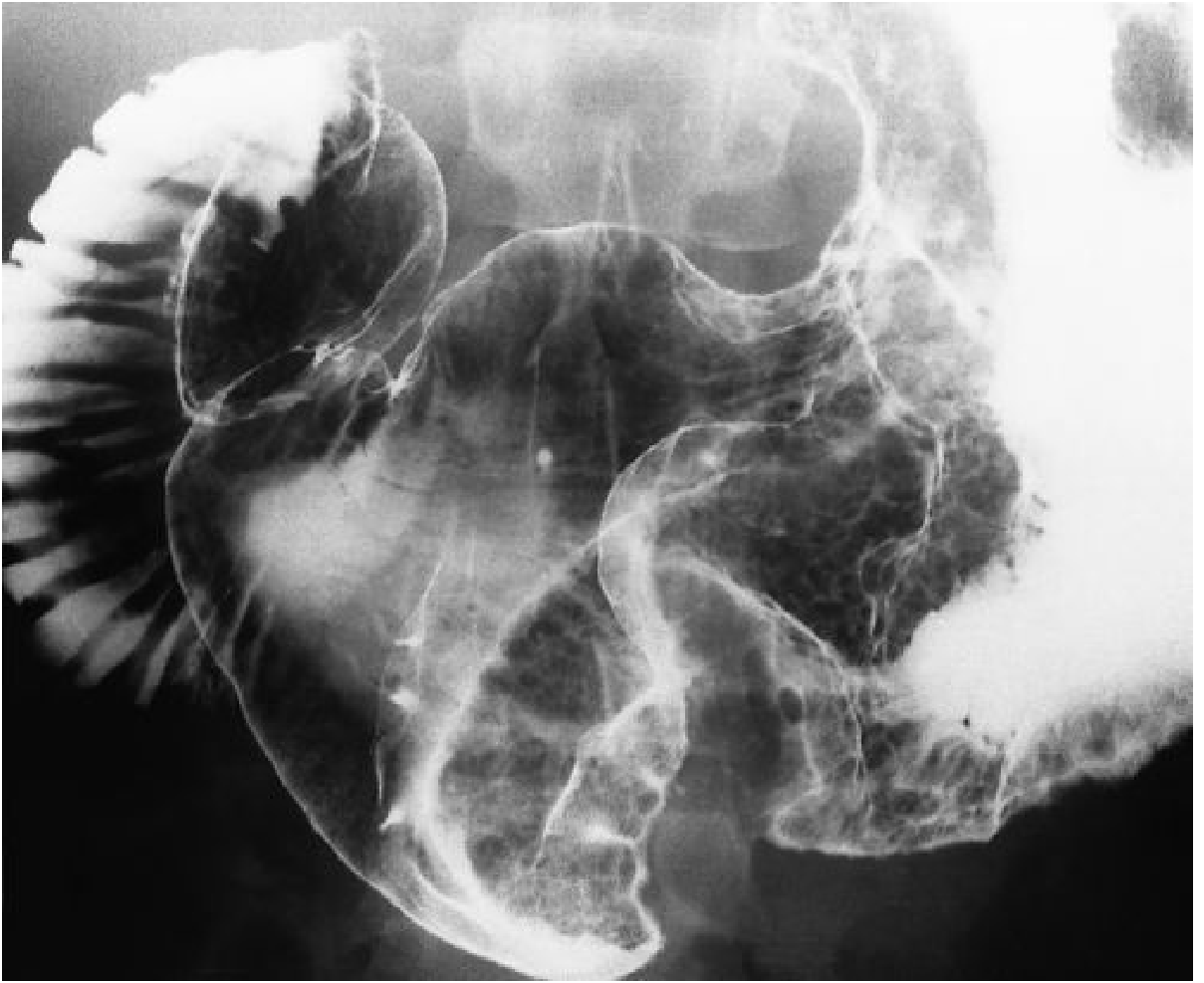
Fig. 20.10 - Grainger & Allison's Diagnostic Radiology: Classic Ménétrier Disease with massively enlarged folds in the body, antrum spared. Note the convoluted, brain-like (cerebriform) appearance of the folds.
Key radiographic features:
- Massively thickened, lobular, often bizarre gastric folds
- Prominent in the proximal stomach and greater curvature
- Antrum generally spared (though involved in up to 50% of cases)
- Folds remain pliable (differentiates from carcinoma, where stomach is rigid/aperistaltic)
- Increased fluid in small bowel may impair optimal barium coating
Histology
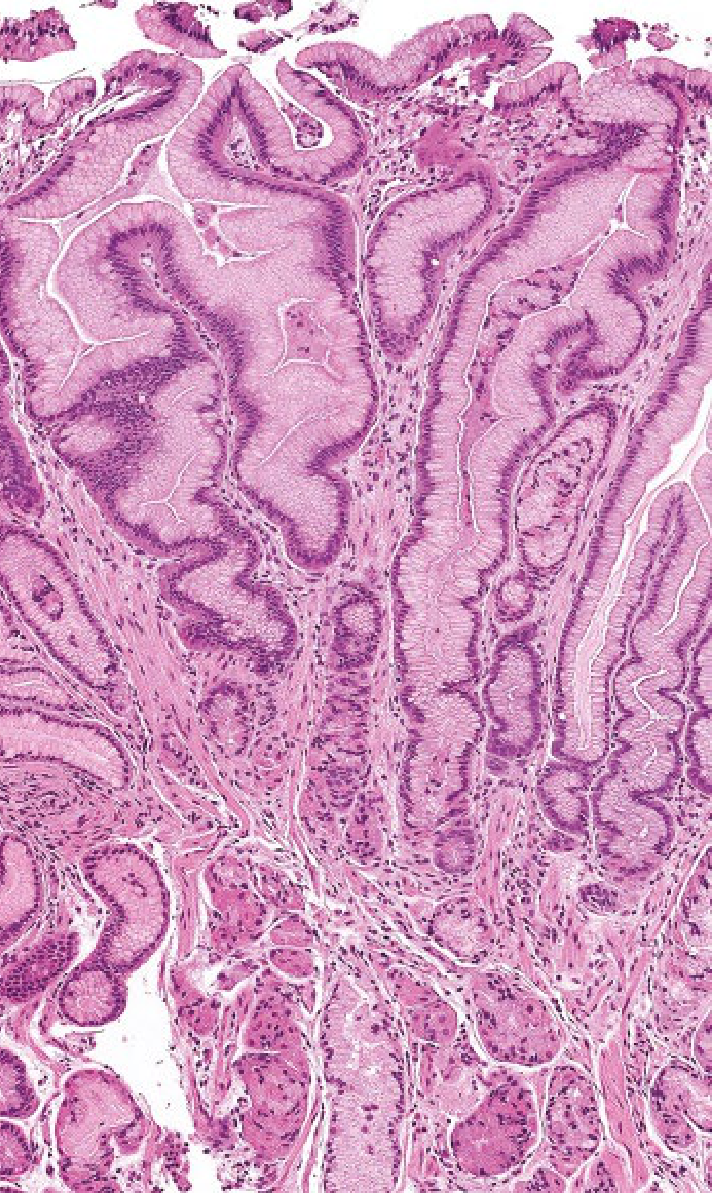
Fig. 51.16 - Yamada's Textbook of Gastroenterology: Massively expanded foveolar epithelium in Ménétrier disease leading to protein loss and hypoalbuminemia
Histological features:
- Massive foveolar hyperplasia - the hallmark
- Glands are elongated with a corkscrew/tortuous appearance
- Cystic gland dilation is common
- Marked reduction in oxyntic glands (parietal + chief cells)
- Inflammation is minimal (not a gastritis - key distinction)
- Intraepithelial lymphocytosis in some cases
Comparison with Other Hypertrophic Gastropathies
| Feature | Ménétrier Disease | Zollinger-Ellison Syndrome |
|---|---|---|
| Location | Body + fundus | Fundus |
| Dominant cell | Mucous (foveolar) | Parietal > mucous |
| Gastric acid | Reduced/absent | Markedly increased |
| Protein loss | Yes | No |
| Associated pathology | TGF-α overexpression | Gastrinoma (MEN-1) |
| Dysplasia/cancer risk | Yes | No |
- Robbins, Cotran & Kumar - Table 17.5
Diagnosis
- Endoscopy with deep mucosal biopsy (snare technique preferred for full-thickness sample)
- Barium upper GI / CT - characteristic giant folds
- Labs: Serum albumin (↓), gastric acid pH (achlorhydria), serum gastrin, CMV and H. pylori serology, CBC
- A nondiagnostic endoscopic biopsy may require surgically obtained full-thickness biopsy to exclude malignancy
Differential diagnosis of giant gastric folds:
- Zollinger-Ellison syndrome
- Gastric lymphoma / infiltrating carcinoma
- CMV / syphilis / tuberculosis / histoplasmosis gastritis
- Sarcoidosis
- Gastric polyposis (FAP, prolonged PPI use)
Treatment
| Approach | Details |
|---|---|
| Supportive | IV albumin infusions, parenteral nutritional supplementation |
| Treat underlying infection | H. pylori eradication, antiviral therapy for CMV |
| Acid suppression | PPIs, H2 blockers (inconsistent benefit) |
| Anti-EGFR therapy | Cetuximab (monoclonal anti-EGFR antibody) - near-complete histologic remission in 4/7 patients in trials - the most targeted therapy |
| Other agents | Octreotide (somatostatin analogue), anticholinergics, corticosteroids |
| Surgery | Gastrectomy for severe, refractory cases; also performed when malignancy cannot be excluded |
In children: disease is usually self-limited; viral clearance (CMV) leads to normalization of epithelial proliferation and restoration of gastric anatomy without surgery.
- Harrison's Principles of Internal Medicine, 22e
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Cancer Risk
MD is considered premalignant in adults - retrospective case-control studies show increased risk of gastric adenocarcinoma with decreased 5- and 10-year survival compared to controls. Long-term endoscopic surveillance is warranted.
Cerebriform nuclei with sezary cells
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Finding Sources
Reading File
All the key content is in hand. Here is the complete answer:
Sézary Syndrome - Cerebriform Nuclei
Cerebriform nuclei (also called cerebroid nuclei) - deeply convoluted, brain-like nuclear infolding visible on peripheral blood smear or skin biopsy - are the pathognomonic feature of Sézary cells, defining Sézary Syndrome.
What Are Sézary Cells?
Sézary cells were first described in 1938 by Albert Sézary as large, atypical, mononuclear cells with lobulated, cerebroid nuclei. Their nucleus has marked infolding of the nuclear membrane, creating a convoluted, brain-like (cerebriform) appearance on light microscopy.
They are malignant CD4+ T helper cells that:
- Home to the skin (via cutaneous lymphocyte antigen / CLA and CCR4)
- Circulate in the peripheral blood (leukemic phase)
- Infiltrate lymph nodes
Sézary Syndrome - Definition
"Sézary syndrome is characterized by the triad of diffuse erythroderma, generalized lymphadenopathy, and circulating malignant T cells with cerebriform nuclei - so-called Sézary cells."
- Fitzpatrick's Dermatology
Sézary Syndrome is the leukemic phase of Cutaneous T-Cell Lymphoma (CTCL) / Mycosis Fungoides (MF), accounting for ~3% of all cutaneous lymphomas.
The Classic Triad
| Component | Details |
|---|---|
| 1. Erythroderma | Generalized, intense fiery-red skin involvement; severe pruritus, burning, episodes of chills |
| 2. Generalized lymphadenopathy | Cervical, axillary, inguinal nodes enlarged |
| 3. Circulating Sézary cells | Malignant T cells with cerebriform nuclei in peripheral blood |
Associated findings:
-
Leonine facies (lion-like face from skin thickening)
-
Eyelid edema / ectropion
-
Diffuse alopecia
-
Hyperkeratosis of palms and soles
-
Dystrophic nails
-
Elevated IgE and eosinophilia (Th2 cytokine phenotype)
-
Andrews' Diseases of the Skin
Peripheral Blood Smear - Sézary Cells
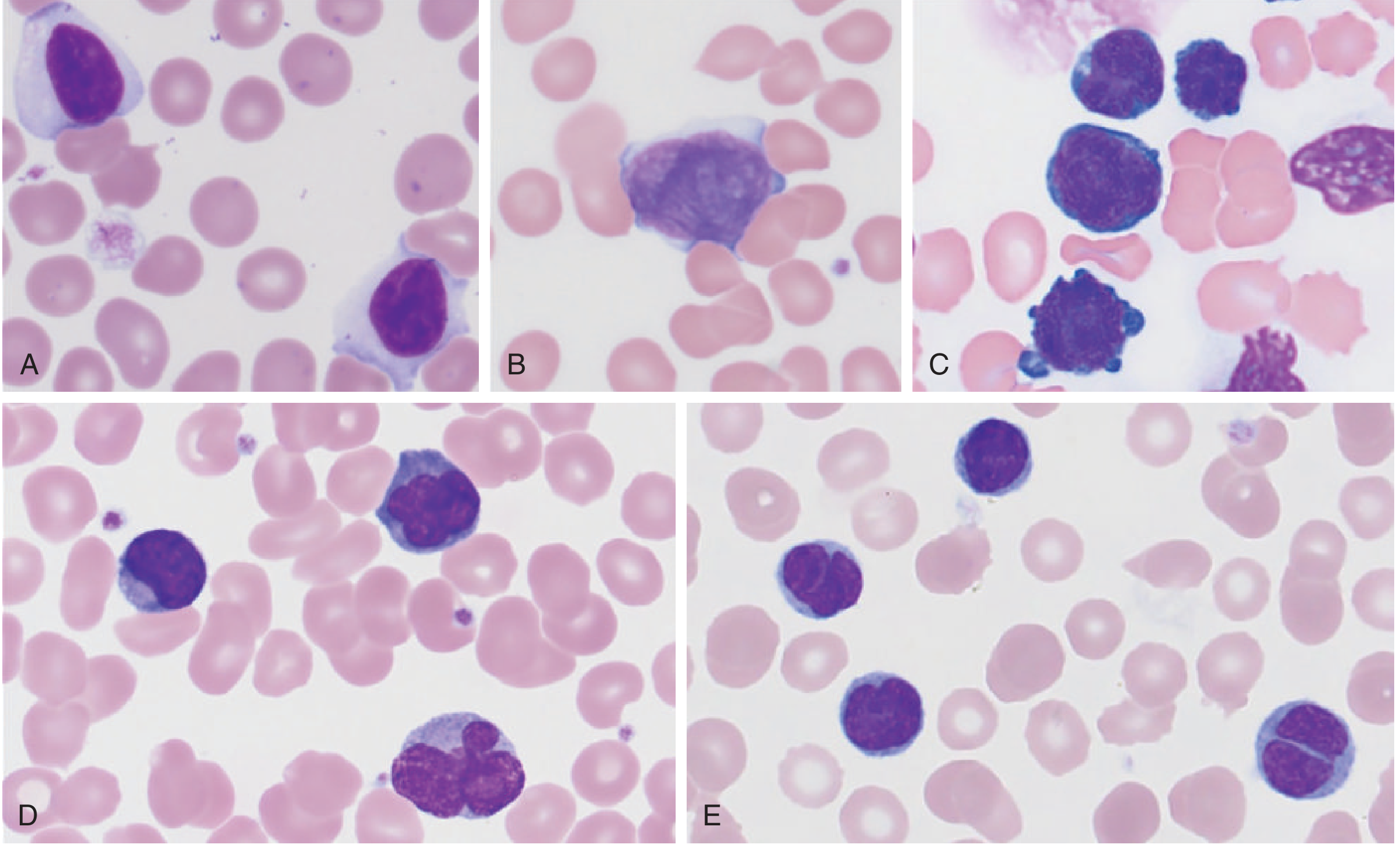
Tietz Textbook of Laboratory Medicine, 7th ed. - Panel B shows the classic Sézary cell with its convoluted "cerebriform" nucleus
Relationship to Mycosis Fungoides (MF)
Sézary Syndrome and Mycosis Fungoides are on a spectrum of CTCL:
Mycosis Fungoides (skin-limited CTCL)
Patch phase → Plaque phase → Tumor phase
↓
Sézary Syndrome (leukemic phase)
[erythroderma + blood involvement]
Key distinction:
- In MF: cutaneous lesions progress through patch → plaque → tumor; skin lesions do proceed to tumefaction
- In Sézary Syndrome: generalized erythroderma from onset; skin lesions rarely proceed to tumefaction; leukemic cells circulate in blood
Both are tumors of CD4+ helper T cells expressing skin-homing molecules (CLA, CCR4). - Robbins, Cotran & Kumar Pathologic Basis of Disease
Histology of MF/Sézary - Skin Biopsy
| Feature | Description |
|---|---|
| Epidermotropism | Neoplastic T cells infiltrate the epidermis (hallmark) |
| Pautrier microabscesses | Clusters of neoplastic T cells within the epidermis - pathognomonic |
| Cerebriform nuclei | Marked nuclear membrane infolding in infiltrating T cells |
| Band-like dermal infiltrate | Lymphocytes in upper dermis with admixed histiocytes, eosinophils |
| Minimal spongiosis | Unlike reactive eczema |
"T cells with irregular nuclei show single-cell infiltration of the epidermis and often form clusters known as Pautrier abscesses... The nuclei of at least a portion of neoplastic cells typically have a cerebriform appearance." - Henry's Clinical Diagnosis and Management by Laboratory Methods
Immunophenotype
| Marker | Sézary Cells |
|---|---|
| CD3 | + |
| CD4 | + (Th2 phenotype) |
| CD8 | - |
| CD7 | Lost (in ~2/3 of cases - key aberrant loss) |
| CD26 | Lost (majority of cases - useful diagnostic marker) |
| CD158k/KIR3DL2 | Aberrantly expressed (normally on NK cells only) |
CD4:CD8 ratio in blood is markedly elevated (>10) - a diagnostic criterion.
Diagnostic Criteria (ISCL/EORTC)
Diagnosis of Sézary Syndrome requires:
- Erythroderma
- Positive T-cell clone in peripheral blood (T-cell receptor gene rearrangement)
- At least one B2 criterion, which includes:
- Absolute Sézary cell count ≥1000 cells/mm³ in peripheral blood
- CD4:CD8 ratio >10
- Loss of CD2, CD3, CD4, or CD5 from circulating T cells
- Lack of CD7 or CD26
Note: Sézary cells alone on cytomorphology are not sufficient - they can be found in other inflammatory erythrodermas (reactive conditions). Clonality must be proven.
Prognosis
| Disease | Median Survival |
|---|---|
| MF (patch/plaque stage) | ~10 years |
| Sézary Syndrome | ~3-5 years (5-year OS: 24-43%) |
Patients often die from infectious complications (reduced cell-mediated immunity due to malignant replacement of normal T cells). Leukocytosis up to 30,000 cells/mm³ can be present.
Treatment Options
| Modality | Notes |
|---|---|
| Low-dose methotrexate | ~50% response rate; median survival ~101 months |
| Extracorporeal photopheresis (ECP) | Photophoresis with combination agents; median survival 39-60 months |
| Total skin electron beam (TSEB) radiation | Complete cutaneous responses; also reduces blood burden |
| PUVA + retinoids + interferon-α | Combination skin-directed/systemic |
| Vorinostat / romidepsin | HDAC inhibitors |
| Mogamulizumab | Anti-CCR4 antibody; approved for relapsed/refractory MF/SS |
| Alemtuzumab | Anti-CD52 |
| Zanolimumab | Anti-CD4 |
- Fitzpatrick's Dermatology; Andrews' Diseases of the Skin
Steatorrhoea with duodenal biopsy Fat cells Acanthocytes
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Finding Sources
Reading File
All the key information is in hand. Here is the complete answer:
Abetalipoproteinemia (Bassen-Kornzweig Syndrome)
The triad of steatorrhoea + fat-laden enterocytes on duodenal biopsy + acanthocytes on peripheral blood smear is the classic presentation of Abetalipoproteinemia - a rare autosomal recessive disorder of lipoprotein assembly.
Definition
Abetalipoproteinemia (ABL) is a rare autosomal recessive disorder caused by mutation in the gene encoding Microsomal Triglyceride Transfer Protein (MTP), resulting in:
-
Complete inability to assemble and secrete triglyceride-rich lipoproteins (chylomicrons, VLDL, LDL)
-
Intracellular lipid accumulation in enterocytes and hepatocytes
-
Severe fat malabsorption with downstream multi-system consequences
-
Robbins & Kumar Basic Pathology; Yamada's Textbook of Gastroenterology, 7e
Pathogenesis - The MTP Defect
Normal pathway:
Dietary fat → Enterocyte → MTP transfers lipids to ApoB48 → Chylomicron → Lymphatics → Blood
ABL:
MTP mutation → Cannot transfer lipids to ApoB48
→ Chylomicrons CANNOT be formed
→ Lipids TRAPPED inside enterocytes (vacuoles)
→ Fat-laden enterocytes on biopsy
→ Steatorrhoea
→ Fat-soluble vitamin deficiency (A, D, E, K)
ApoB isoforms affected:
| Isoform | Site | Lipoprotein formed | In ABL |
|---|---|---|---|
| ApoB48 | Enterocytes (edited mRNA) | Chylomicrons | ABSENT |
| ApoB100 | Hepatocytes (unedited mRNA) | VLDL, LDL | ABSENT |
Both are absent because the defect is in MTP, which is required for both. - Yamada's Textbook of Gastroenterology, 7e
The Three Key Diagnostic Clues Explained
1. Steatorrhoea
Fat cannot be exported from enterocytes (no chylomicrons), so dietary fat is not absorbed. Fat passes through in the stool. Infants present with diarrhea, vomiting, bloating, and failure to thrive from birth. Breast milk (high in fat) is poorly tolerated.
2. Duodenal Biopsy - Fat-Laden Enterocytes
Endoscopic examination of the duodenum and jejunum reveals a "white frosting" appearance coating the mucosa from lipid-filled villi. On histology:
- Enterocytes packed with lipid-filled cytoplasmic vacuoles (fat droplets trapped inside)
- Highlighted by Oil Red O stain (especially prominent after a fatty meal)
- Villous architecture is preserved (distinguishes from coeliac disease)
"In the intestine, endoscopic examination reveals a white frosting appearance coating the duodenum and jejunum due to the lipid content in the mucosa. On histological examination, the enterocytes are filled with numerous lipid-filled cytoplasmic vacuoles." - Yamada's Textbook of Gastroenterology, 7e
3. Acanthocytes (Spur Cells)
Because essential fatty acids cannot be absorbed and transported, the lipid composition of red blood cell membranes is altered. This produces acanthocytes - RBCs with irregular, spiky membrane protrusions - visible on peripheral blood smear.
"Failure to absorb essential fatty acids leads to deficiencies of fat-soluble vitamins as well as lipid membrane defects that can be recognized by the presence of acanthocytes (red cells with spiky membrane protrusions) in peripheral blood smears." - Robbins, Cotran & Kumar Pathologic Basis of Disease
Full Clinical Picture
Presentation timeline:
| Age | Manifestations |
|---|---|
| Infancy | Failure to thrive, diarrhea, steatorrhoea, vomiting, abdominal distension |
| Childhood | Fat-soluble vitamin deficiencies become clinically apparent |
| 1st-2nd decade | Neurological + retinal complications if untreated |
| Late | Cardiac complications (cardiomyopathy) |
Multi-System Complications (from Fat-Soluble Vitamin Deficiency)
| Deficiency | Consequence |
|---|---|
| Vitamin E (most severely depleted - no alternative transport) | Spinocerebellar degeneration, peripheral neuropathy, retinitis pigmentosa |
| Vitamin A | Night blindness, retinal degeneration |
| Vitamin K | Coagulopathy (prolonged PT) |
| Vitamin D | Metabolic bone disease |
| Essential fatty acids | Acanthocytosis, membrane instability |
Neurological Syndrome (Bassen-Kornzweig)
Progressive and severe if untreated - develops in the first two decades of life:
- Peripheral neuropathy - earliest sign; areflexia, loss of proprioception/vibration (large-fibre), gait ataxia
- Spinocerebellar degeneration - cerebellar ataxia of gait, trunk, extremities; dysarthria; titubation
- Retinitis pigmentosa - progressive visual field constriction, ring scotomata
- Ophthalmoplegia (late)
- Skeletal deformity - pes cavus, kyphoscoliosis (secondary to early neuropathy)
- Cardiomyopathy - cardiac enlargement, congestive failure (serious late complication)
"Most untreated patients develop retinitis pigmentosa, peripheral neuropathy, and spinocerebellar degeneration during the first two decades of life." - Bradley and Daroff's Neurology in Clinical Practice
Laboratory Findings
| Test | Result |
|---|---|
| Serum cholesterol | Markedly low (20-50 mg/dL; normal >150) |
| Triglycerides (fasting) | <10 mg/dL |
| Post-fat-load triglycerides | No rise (diagnostic) |
| Post-fat-load chylomicrons | Absent (diagnostic) |
| Plasma ApoB48, ApoB100 | Absent |
| Vitamin E | Extremely low |
| Vitamins A, D, K | Low (but less severely) |
| Peripheral smear | Acanthocytes |
Diagnosis Summary
| Feature | Finding |
|---|---|
| Genetics | Autosomal recessive; MTP gene mutation; consanguinity common |
| Endoscopy | White frosting of duodenum/jejunum |
| Duodenal biopsy | Lipid-vacuolated enterocytes (Oil Red O +); normal villous architecture |
| Blood smear | Acanthocytes (spur cells) |
| Lipid panel | Absent ApoB, absent chylomicrons, hypocholesterolaemia |
| Neuroimaging | Spinocerebellar atrophy in advanced cases |
Treatment
| Intervention | Details |
|---|---|
| Dietary fat restriction | <15 g fat/day - minimizes steatorrhoea |
| Medium-chain triglycerides (MCT) | Absorbed directly without chylomicron packaging - provides calories |
| Vitamin E supplementation | 100 mg/kg/day (vs. normal 10-30 mg/day) - prevents/arrests neurological complications. If started early (before 16 months), neurological and retinal symptoms do not develop |
| Vitamins A, K, D | Standard supplementation; adequate levels achievable |
Key point: Early treatment with high-dose vitamin E can prevent neurological deterioration entirely. Late treatment can halt or partially reverse progression. - Yamada's Textbook of Gastroenterology, 7e
Carneys triad and carneys complex
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
All the essential content is in hand. Here is the complete side-by-side breakdown:
Carney Triad vs. Carney Complex - A Critical Distinction
These two are completely separate entities - different tumors, different genetics, different inheritance, different eponyms (both named after Dr. J. Aidan Carney, a Mayo Clinic pathologist/endocrinologist).
1. Carney Triad
Definition
First described in the 1970s. A sporadic (non-familial, non-hereditary) syndrome defined by the co-occurrence of three tumor types:
| Component | Tumor |
|---|---|
| 1 | Gastric GIST (Gastrointestinal Stromal Tumor) - often multifocal, with lymph node involvement |
| 2 | Extra-adrenal Paraganglioma |
| 3 | Pulmonary Chondroma |
All three need not be present simultaneously - the triad may be incomplete and evolve over time.
An expanded description also includes adrenal adenomas and esophageal leiomyomas in some patients. - Sleisenger & Fordtran's GI and Liver Disease
Key Features
| Feature | Detail |
|---|---|
| Inheritance | Sporadic - NOT familial, NOT inherited |
| Gender | Predominantly young females |
| Age | Young patients (often <30 years at presentation) |
| Genetics | Unknown cause - NOT associated with SDH gene mutations (unlike Carney-Stratakis) |
| GIST molecular profile | KIT/PDGFRA mutation-negative; SDHB-negative by IHC |
| Imatinib response | Generally ineffective (KIT/PDGFRA wild-type) |
"The Carney triad is a syndrome of unknown cause defined by the co-occurrence of GIST, paraganglioma, and pulmonary chondroma, usually occurring in females." - Robbins, Cotran & Kumar Pathologic Basis of Disease
Carney Triad vs. Carney-Stratakis Dyad (Carney-Stratakis Syndrome)
These are often confused - an important distinction:
| Carney Triad | Carney-Stratakis Dyad | |
|---|---|---|
| Components | GIST + Paraganglioma + Pulmonary chondroma | GIST + Paraganglioma (only 2) |
| Inheritance | Sporadic | Familial (autosomal dominant) |
| Genetics | SDH mutation absent | SDH gene mutations (SDHA/B/C/D germline) |
| SDHB IHC | Negative | Negative |
| Family history | Absent | Present - family members also affected |
"Evaluation has identified that tumors part of the Dyad carry mutations in the SDH family of genes, whereas those associated with the triad do not have such mutations." - Sleisenger & Fordtran's GI and Liver Disease
Clinical implication: Any patient with gastric GIST lacking SDHB expression on IHC requires genetic counseling to distinguish between Carney Triad (sporadic) and Carney-Stratakis Dyad (heritable SDH mutation - family screening needed).
2. Carney Complex
Definition
First described in 1985. An autosomal dominant multiple neoplasia syndrome involving myxomas, skin pigmentation, and endocrine tumors.
Also known historically as:
- NAME syndrome: Nevi, Atrial myxoma, Myxoid neurofibromas, Ephelides
- LAMB syndrome: Lentigines, Atrial myxoma, Mucocutaneous myxomas, Blue nevi
The Components
| Feature | Frequency |
|---|---|
| Cardiac myxomas | 79% - any of 4 chambers; recurrent in 20%; can embolize |
| Spotty skin pigmentation (lentigines, blue nevi - epithelioid type) | 65% |
| Primary Pigmented Nodular Adrenocortical Disease (PPNAD) → Cushing syndrome | 45% |
| Cutaneous myxomas (<1 cm papules; ears, eyelids, nipples) | 45% |
| Testicular tumors (large-cell calcifying Sertoli cell tumors → sexual precocity/gynecomastia) | 56% of males |
| Mammary myxoid fibromas | 30% |
| Pituitary somatotrope adenoma → Acromegaly | ~10% |
| Psammomatous melanotic schwannomas (sympathetic chain, upper GI) | Present |
| Thyroid tumors, ovarian cysts | Variable |
"Carney complex is an autosomal dominant disorder characterized by spotty skin pigmentation, myxomas (heart, skin, breast), psammomatous melanotic schwannomas, and endocrine tumors involving the adrenals, Sertoli cells, somatotropes, thyroid, and ovary." - Harrison's Principles of Internal Medicine, 22e
Genetics
| Type | Gene | Chromosome |
|---|---|---|
| CNC Type 1 (majority, ~50%) | PRKAR1A (Protein Kinase A Regulatory Subunit 1α) - tumour suppressor | 17q22-24 |
| CNC Type 2 | Unknown gene | 2p16 |
- PRKAR1A is a tumour suppressor - mutations cause loss of inhibition of PKA (protein kinase A) signalling, leading to uncontrolled cell proliferation
Key Clinical Points for Carney Complex
- Cardiac myxomas are life-threatening - can embolize, causing stroke or peripheral ischaemia. Recognition of the syndrome and surgical removal is lifesaving
- Cushing syndrome in Carney complex is atypical - patients are often thin (not obese) with normal or only mildly elevated urinary free cortisol; may show periodic/cyclical Cushing's (fluctuating cortisol production)
- Pigmentation distribution is characteristic - vermilion border of lips, lacrimal caruncle, conjunctival semilunar fold, sclera (not perioral like Peutz-Jeghers)
- Cardiac myxomas are recurrent (20%) and multichambered (unlike sporadic myxomas which are usually solitary left atrial)
- Screen first-degree relatives - autosomal dominant inheritance
- Andrews' Diseases of the Skin; Fitzpatrick's Dermatology
Side-by-Side Comparison Table
| Feature | Carney TRIAD | Carney COMPLEX |
|---|---|---|
| Year described | 1970s | 1985 |
| Nature | Tumour syndrome | Neoplasia + pigmentation + endocrine syndrome |
| Inheritance | Sporadic | Autosomal dominant |
| Gene | Unknown (not SDH) | PRKAR1A (17q) |
| Key tumours | GIST + Paraganglioma + Pulmonary chondroma | Cardiac myxoma + Endocrine tumours + Schwannoma |
| Gender predominance | Young females | Either sex |
| Life-threatening lesion | Paraganglioma | Cardiac myxoma (embolism) |
| Skin findings | None specific | Lentigines, blue nevi (lips, conjunctiva) |
| Cushing syndrome | No | Yes (PPNAD) - atypical (thin patient) |
| SDH mutation | Absent | Not relevant |
| Family screening | Not needed | Mandatory (AD inheritance) |
| GIST molecular type | KIT/PDGFRA wild-type, SDHB-negative | Not a feature |
Hepatitis c infection with councilman bodies
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Loading Image
I have all the key content needed. Here is the complete, well-structured answer:
Hepatitis C - Councilman Bodies / Acidophil Bodies
This question hinges on a critical terminological distinction in liver pathology. Understanding it is essential for exams and clinical practice.
The Terminology - Councilman Body vs. Acidophil Body
This is one of the most tested distinctions in hepatic pathology:
| Term | Used in | Meaning |
|---|---|---|
| Councilman body | Yellow fever only | Apoptotic hepatocyte first described by William Thomas Councilman in yellow fever (1890) |
| Acidophil body | All other forms of hepatitis (including HCV) | The same morphological entity - apoptotic hepatocyte - but called "acidophil body" by convention in viral hepatitis, drug-induced liver injury, etc. |
"Apoptotic hepatocytes were first clearly described in yellow fever by William Thomas Councilman and therefore have been referred to as Councilman bodies; while apoptosis occurs in many forms of liver disease, by convention this eponym is restricted to that disease. In other settings in which apoptotic hepatocytes are seen (e.g., acute and chronic hepatitis), the term acidophil bodies is used, due to their deeply eosinophilic staining characteristics."
- Robbins, Cotran & Kumar Pathologic Basis of Disease
So the correct terminology in Hepatitis C is acidophil bodies, not Councilman bodies - though they are morphologically identical.
What Is an Acidophil / Councilman Body?
It is a hepatocyte undergoing apoptosis (programmed cell death):
Hepatocyte under immune attack (CD8+ T cells, cytokines)
↓
Caspase activation → Programmed cell death
↓
Hepatocyte SHRINKS (opposite of necrosis which swells)
↓
Nuclear chromatin condensation (pyknosis)
↓
Nuclear fragmentation (karyorrhexis)
↓
Cell fragments into small, densely eosinophilic globules
↓
ACIDOPHIL BODY (deeply pink/eosinophilic, anucleate or with pyknotic nucleus)
These round, shrunken, deeply eosinophilic (acidophilic) apoptotic bodies are extruded into the sinusoids or remain in the hepatic parenchyma and are rapidly phagocytosed.
Hepatitis C Liver Biopsy - Histological Features
The liver biopsy in chronic HCV infection has characteristic histological findings. Acidophil bodies are one component of the overall picture:
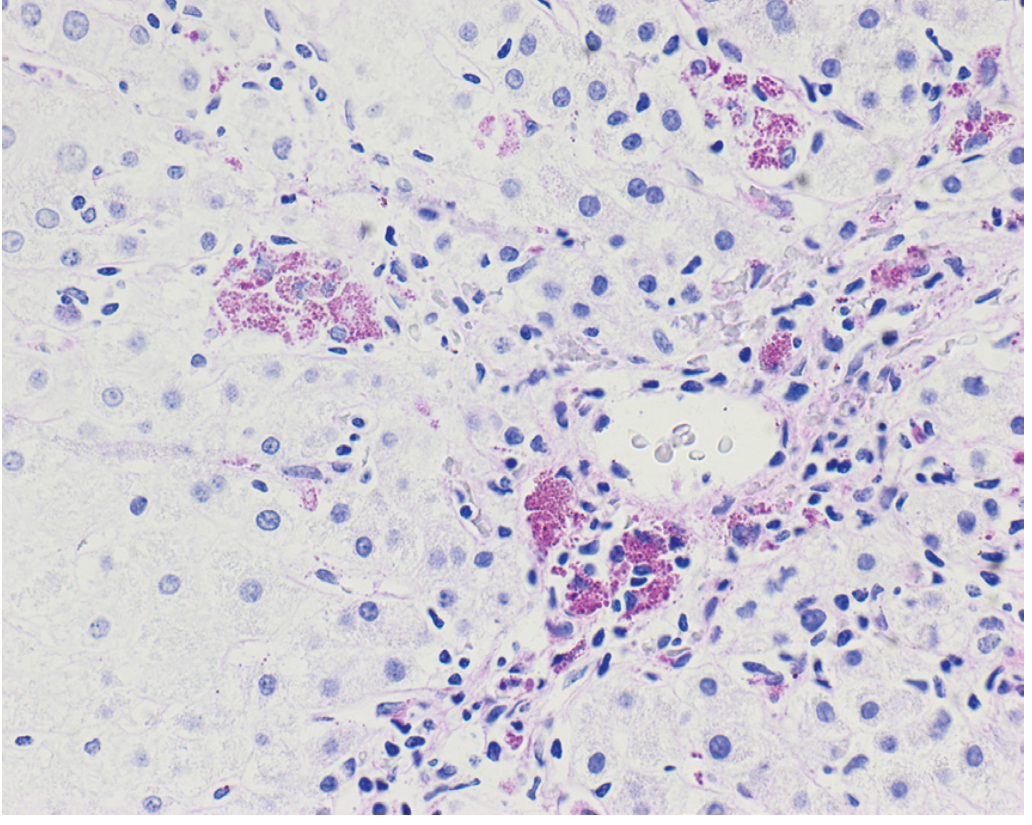
Fig. 14.3 - Robbins & Kumar Basic Pathology: Biopsy from a patient with chronic hepatitis C showing acidophil bodies (single arrows) and patchy inflammatory infiltrate (double arrows)
Complete Histological Picture of Chronic HCV
| Feature | Description |
|---|---|
| Acidophil bodies | Scattered apoptotic hepatocytes - shrunken, deeply eosinophilic, anucleate; hallmark of ongoing hepatocyte death |
| Portal tract lymphoid aggregates | Dense lymphoid follicle-like aggregates in portal tracts - characteristic of HCV (less common in HBV) |
| Bile duct damage | Lymphocytic infiltration and damage of interlobular bile ducts ("biliary injury") - HCV-specific |
| Steatosis | Macrovesicular fat - particularly prominent in HCV genotype 3 (direct viral steatosis); also in other genotypes (metabolic steatosis) |
| Interface hepatitis | Lymphocytic spillover from portal tracts into surrounding periportal hepatocytes ("piecemeal necrosis") |
| Lobular inflammation | Scattered intralobular lymphocytes and Kupffer cell activation |
| Sinusoidal lymphocytosis | Single-file lymphocytes in sinusoids ("string of beads") |
| Perisinusoidal/pericellular fibrosis | With progressive disease |
| Cirrhosis | End-stage - nodular regeneration with bridging fibrosis |
Note - what is NOT seen in HCV (unlike HBV):
- No "ground glass" hepatocytes - Ground glass cells (HBsAg-laden smooth ER) are specific to HBV, not HCV
Mechanism of Hepatocyte Death in HCV
The immune response is the primary driver:
HCV RNA replication in hepatocytes
↓
Viral antigens presented on MHC Class I
↓
CD8+ cytotoxic T lymphocytes recognize and attack infected hepatocytes
↓
Perforin/granzyme pathway → Caspase activation → APOPTOSIS
Fas/FasL pathway → Caspase activation → APOPTOSIS
↓
Acidophil bodies form
↓
Hepatic stellate cell activation by cytokines (TGF-β, TNF-α)
↓
Perisinusoidal myofibroblasts → Collagen deposition → FIBROSIS → CIRRHOSIS
"Adaptive immune cells play a critical role in viral hepatitis, with CD4+ and CD8+ T cells being particularly important in the eradication of virus-infected hepatocytes and in causing liver injury in chronic disease." - Robbins & Kumar Basic Pathology
Summary - Key Points for Exams
| Point | Fact |
|---|---|
| Councilman bodies = Yellow fever only | First described by W.T. Councilman, 1890 |
| Same structure in HCV = Acidophil bodies | Named for deep eosinophilic staining |
| Mechanism | Hepatocyte apoptosis (NOT necrosis) |
| Morphology | Shrunken, deeply eosinophilic, pyknotic/anucleate globules |
| HCV-specific histological features | Portal lymphoid aggregates + bile duct damage + steatosis + acidophil bodies |
| Ground glass cells | HBV only (NOT HCV) |
| Fibrosis driver | Hepatic stellate cell → myofibroblast → collagen |
This is a shared conversation. Sign in to Orris to start your own chat.