Explain to me pathophys of Aortic regurgitation
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have all the content I need from Braunwald's Heart Disease. The pathophysiology section was truncated but I captured the key content. Let me compile the comprehensive answer now.
Here is a detailed explanation of the pathophysiology of Aortic Regurgitation (AR), drawing from Braunwald's Heart Disease:
Pathophysiology of Aortic Regurgitation
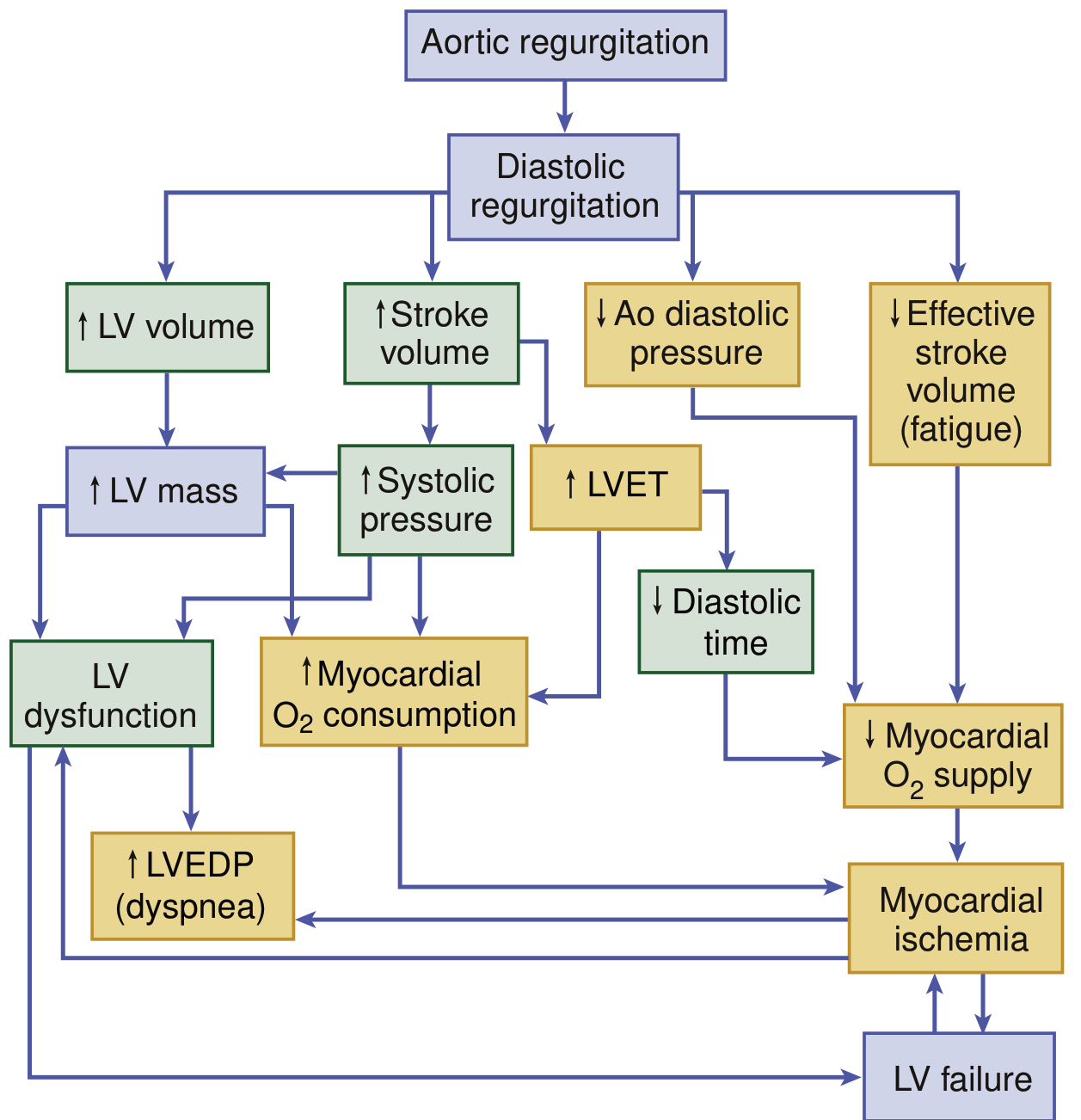
The Core Defect: Diastolic Regurgitation
In AR, the aortic valve fails to close competently during diastole, allowing blood to flow back from the aorta into the left ventricle (LV). This creates a volume overload state that is distinct from the pressure overload of aortic stenosis.
The regurgitant volume depends on:
- The size of the regurgitant orifice
- The duration of diastole (so slower heart rates = more regurgitation per beat)
- The diastolic pressure gradient between the aorta and LV
Chronic Aortic Regurgitation
1. Volume Overload and LV Dilation (Eccentric Hypertrophy)
The LV receives its normal pulmonary venous inflow plus the regurgitant volume returning from the aorta. Over time, this combined volume load causes:
- Increased LV end-diastolic volume (LVEDV) - the LV dilates progressively
- Eccentric hypertrophy - new sarcomeres are added in series, so the LV cavity enlarges without proportionate wall thickening (unlike the concentric hypertrophy of pressure overload)
- Increased total stroke volume - the LV ejects a larger-than-normal stroke volume to compensate, maintaining forward cardiac output despite the regurgitant fraction
The ratio of LV cavity radius to wall thickness increases, which by the Law of Laplace (wall stress = pressure × radius / 2 × wall thickness) increases systolic wall stress. This stimulates further hypertrophy to normalize wall stress - a compensatory but ultimately failing mechanism.
2. Hemodynamic Consequences - The Wide Pulse Pressure
- Increased systolic BP: The large stroke volume ejected into the aorta raises systolic pressure
- Decreased diastolic BP: Continuous back-leak of blood lowers aortic diastolic pressure
- The result is a wide pulse pressure (e.g., 160/50 mmHg), the hallmark of chronic severe AR
This produces the characteristic peripheral signs:
- Water-hammer (Corrigan) pulse - bounding, collapsing
- De Musset sign (head bobbing)
- Quincke's pulses (nail bed pulsations)
- Duroziez sign (femoral artery murmur)
- Hill's sign (leg systolic BP > arm systolic BP by >20 mmHg)
3. Myocardial Oxygen Supply-Demand Mismatch
This is one of the most clinically important aspects of AR pathophysiology:
Increased O2 demand from:
- Increased LV mass (hypertrophy)
- Increased LV wall stress
- Prolonged LV ejection time (LVET) - the LV spends more time in systole ejecting its larger stroke volume, reducing diastolic filling time
Decreased O2 supply from:
- Low aortic diastolic pressure reduces coronary perfusion pressure (coronary filling predominantly occurs during diastole)
- Reduced diastolic filling time (as LVET increases, diastole is shortened)
- Increased LV end-diastolic pressure (LVEDP) compresses subendocardial coronary vessels
The net result is subendocardial ischemia, which can cause angina even in the absence of epicardial coronary artery disease. Studies have confirmed reduced coronary flow reserve in chronic severe AR.
4. Compensated vs. Decompensated Phases
Compensated phase (asymptomatic):
- The LV dilates and hypertrophies to accommodate the volume load
- Ejection fraction (EF) is maintained through the Frank-Starling mechanism
- Patients may remain asymptomatic for years to decades
- Forward cardiac output is preserved
Decompensated phase:
- After prolonged volume overload, the LV myocardium undergoes fibrosis and cellular dysfunction
- Preload reserve is exhausted - the LV cannot further dilate to compensate
- EF falls, first subtly (EF 50-55%) then overtly (EF <50%)
- LVEDP rises, causing pulmonary congestion and dyspnea
- The LV progressively dilates further, worsening functional MR
- Symptoms emerge: exertional dyspnea, orthopnea, paroxysmal nocturnal dyspnea
- Angina and ultimately heart failure develop
A key clinical point: LV dysfunction may become irreversible if AVR is delayed too long after decompensation begins, making early identification of the compensated-to-decompensated transition critical.
Acute Aortic Regurgitation
Acute AR (e.g., from aortic dissection, acute infective endocarditis, or chest trauma) has a distinctly different and more dangerous pathophysiology:
- The LV has no time to dilate and compensate - it is a normal-sized ventricle suddenly receiving a large regurgitant volume
- LVEDP rises acutely and dramatically
- The sudden rise in LVEDP may exceed the pulmonary capillary wedge pressure, causing acute pulmonary edema
- The mitral valve may close prematurely in diastole (due to very high LVEDP) - this is an important echocardiographic sign of acute severe AR
- Forward cardiac output falls precipitously - cardiogenic shock
- Tachycardia develops as a compensatory response (and is actually helpful here, as it shortens diastole and reduces regurgitant time)
- Heart sounds are characteristically soft (LV is not dilated, so less pounding); peripheral signs like water-hammer pulse may be absent despite severe AR
- This is a cardiac emergency requiring urgent surgical intervention
Summary Table
| Feature | Chronic AR | Acute AR |
|---|---|---|
| LV size | Markedly enlarged | Normal |
| LV compliance | Increased | Normal/reduced |
| LVEDP | Normal early, rises late | Markedly elevated |
| Pulse pressure | Wide | May be normal |
| Peripheral signs | Prominent | Often absent |
| Heart sounds | Hyperdynamic | Soft |
| Compensation | Eccentric hypertrophy | None (no time) |
| Urgency | Elective monitoring then surgery | Emergency surgery |
Sources: Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine, Chapter 73; Fuster and Hurst's The Heart, 15th Edition.
This is a shared conversation. Sign in to Orris to start your own chat.