I now have comprehensive, authoritative content from multiple textbooks plus key images. Here is the complete synthesis:
Hemolytic Jaundice — Comprehensive Clinical Review
1. Normal Bilirubin Physiology
Understanding hemolytic jaundice begins with the normal pathway of bilirubin metabolism.
Step-by-step process:
-
RBC senescence: After ~120 days, aged RBCs are phagocytized by macrophages of the mononuclear phagocyte system (MPS) — primarily in the spleen, liver, and bone marrow. (~85% of heme comes from RBC breakdown; the remaining 15% from other heme proteins such as myoglobin and cytochromes.)
-
Heme catabolism: Hemoglobin is split into globin + heme. Heme oxygenase (in macrophages) oxidizes heme, opening the porphyrin ring → biliverdin (green) + CO + Fe²⁺. Biliverdin reductase then reduces biliverdin → unconjugated bilirubin (UCB) (indirect bilirubin), a red-orange, lipid-soluble pigment.
-
Transport in blood: UCB is poorly soluble in plasma, so it binds tightly to albumin for transport to the liver. (Note: Drugs like salicylates and sulfonamides can displace bilirubin from albumin, risking neural toxicity in neonates.)
-
Hepatic uptake: UCB dissociates from albumin, enters hepatocytes via facilitated diffusion (OATP transporters), and binds to intracellular carrier proteins, especially ligandin (glutathione-S-transferase).
-
Conjugation: In the smooth ER, bilirubin UDP-glucuronosyltransferase (UGT1A1) adds two glucuronic acid molecules → bilirubin diglucuronide (conjugated bilirubin, CB / direct bilirubin). CB is water-soluble.
-
Biliary excretion: CB is actively secreted into bile canaliculi via the MRP2 (ABCC2) transporter → enters bile → small intestine.
-
Intestinal conversion: Colonic bacteria deconjugate CB and reduce it to urobilinogen (colorless). Most is excreted in stool, becoming oxidized to stercobilin (brown color of stool).
-
Enterohepatic circulation: ~20% of urobilinogen is reabsorbed into the portal circulation, taken up by the liver, and re-excreted into bile. A small fraction (~5%) escapes into systemic circulation and is excreted by the kidneys as urobilin (giving urine its yellow color).
Normal values: Total serum bilirubin ≈ 0.5 mg/dL (range 0.2–1.2 mg/dL), predominantly indirect (UCB).
— Biochemistry, 8th ed, Lippincott Illustrated Reviews, p. 794–797; Guyton and Hall Textbook of Medical Physiology, p. 862–863
2. Mechanism of Hemolytic Jaundice (Pathogenesis)
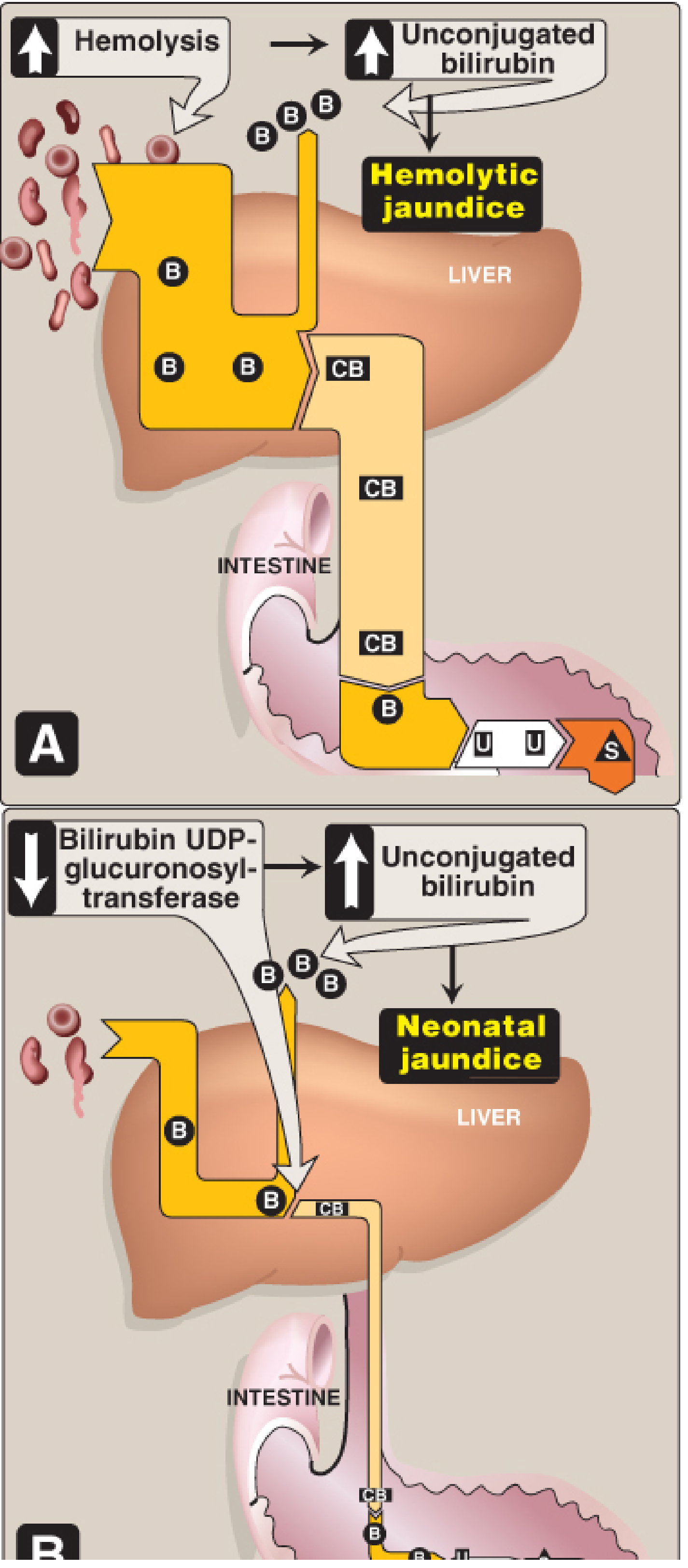
Figure 21.12A — Hemolytic jaundice. Increased hemolysis floods the liver with bilirubin (B), generating excess conjugated bilirubin (CB) and maximizing urobilinogen (U) and stercobilin (S). Only UCB is abnormally elevated. — Lippincott Biochemistry, 8th ed
Hemolytic jaundice is a pre-hepatic (overproduction) type of jaundice. The liver's conjugation capacity is intrinsically normal but is overwhelmed:
- Accelerated RBC destruction releases massive amounts of hemoglobin → increased heme catabolism → excess UCB production.
- The liver, even at maximum capacity, cannot conjugate and excrete bilirubin as rapidly as it is formed.
- Plasma UCB rises (unconjugated hyperbilirubinemia). The liver handles the load until bilirubin production exceeds hepatic conjugation capacity (ceiling ~4 mg/dL from hemolysis alone in a normal liver).
- CB levels may reach the upper range of normal hepatic capacity; large amounts are secreted into bile.
- Urobilinogen formation is markedly increased, as more CB reaches the intestine → more enterohepatic cycling → urinary urobilinogen increased.
- Bilirubin is NOT found in urine (bilirubinuria absent), because UCB is bound to albumin and cannot pass the glomerular filter. The combination of increased urinary urobilinogen + absent urinary bilirubin is the hallmark of hemolytic jaundice.
- Stools are dark (excess stercobilin).
— Guyton and Hall, p. 863; Lippincott Biochemistry, p. 800–801; Harper's Illustrated Biochemistry, 32nd ed
3. Etiology
A. Intravascular Hemolysis (RBC destruction within blood vessels)
| Cause | Mechanism |
|---|
| G6PD deficiency | Lack of NADPH → oxidative stress → Heinz body formation → RBC destruction; triggered by infections, fava beans, primaquine, dapsone |
| Paroxysmal nocturnal hemoglobinuria (PNH) | Clonal GPI-anchor deficiency → complement-mediated lysis (CD55/CD59 absent) |
| Microangiopathic hemolytic anemia (MAHA) — TTP, HUS, DIC | Mechanical shearing of RBCs by fibrin strands → schistocytes |
| Malaria (P. falciparum) | Direct parasitic rupture of RBCs + immune-mediated hemolysis; "blackwater fever" in severe cases |
| ABO-incompatible transfusion | IgM-mediated complement activation → acute intravascular hemolysis |
| Cold agglutinin disease | IgM autoantibodies activate complement at cold extremities |
B. Extravascular Hemolysis (RBC destruction in spleen/liver)
| Cause | Mechanism |
|---|
| Hereditary spherocytosis (HS) | Defects in spectrin, ankyrin, band 3, protein 4.2 → loss of RBC membrane → spherocytes → splenic trapping & phagocytosis |
| Hereditary elliptocytosis (HE) | Spectrin/protein 4.1 defects → rigid elliptical cells |
| Sickle cell disease (HbS) | Polymerization of HbS → rigid, sickle-shaped cells → vaso-occlusion + splenic phagocytosis |
| Beta-thalassemia major | Excess alpha chains precipitate → ineffective erythropoiesis + hemolysis |
| Autoimmune hemolytic anemia (AIHA) — Warm type | IgG autoantibodies (often anti-Rh) coat RBCs → Fc receptor-mediated phagocytosis in spleen |
| Hemolytic disease of the newborn (HDN) | Maternal IgG anti-D crosses placenta → fetal RBC destruction |
| Drug-induced hemolysis | Hapten mechanism (penicillin), immune complex (quinidine), or autoantibody induction (methyldopa) |
| Hypersplenism | Enlarged spleen sequesters and destroys normal RBCs |
— Robbins & Kumar Pathology; Harrison's Principles of Internal Medicine, 22nd ed
4. Pathology
Extravascular hemolysis (most common):
- Spleen: Congestion and enlargement (splenomegaly); hyperplasia of red pulp macrophages, which are engorged with hemosiderin and RBC debris.
- Liver (Kupffer cells): Also participate in phagocytosis of abnormal RBCs; hepatic sinusoidal dilatation.
- Bone marrow: Marked erythroid hyperplasia (compensatory); in chronic hemolysis, extramedullary hematopoiesis occurs (in spleen, liver).
- Gallbladder: Pigment (bilirubin) gallstones — a pathological hallmark of chronic hemolytic states.
- Blood: Peripheral smear shows the causative morphology (spherocytes in HS, schistocytes in MAHA, sickled cells in SCD, target cells in thalassemia).
Intravascular hemolysis additionally shows:
- Hemoglobinemia (pink plasma)
- Hemoglobinuria (dark/red urine)
- Hemosiderinuria (chronic intravascular hemolysis, e.g., PNH)
- Renal tubular damage in severe cases (acute kidney injury)
5. Pathogenesis Leading to Clinical Symptoms
Accelerated RBC destruction
↓
↑ Free Hgb released → Spleen/liver macrophages phagocytize → ↑ UCB production
↓ ↓
(Intravascular): Liver overwhelmed
Hgb → binds haptoglobin → Unconjugated hyperbilirubinemia
(haptoglobin depleted) ↓
Hgb → filtered by kidney JAUNDICE (yellow skin/sclera)
→ hemoglobinuria (dark urine) ↓
↓ ↑ CB in bile → ↑ urobilinogen
↓ → dark stools, ↑ urinary urobilinogen
Compensatory ↑ erythropoiesis
→ reticulocytosis, bone marrow hyperplasia
→ splenomegaly (extramedullary hematopoiesis + RBC destruction)
→ ANEMIA (if destruction > production)
→ pallor, fatigue, tachycardia, dyspnea
6. Clinical Features & Symptoms
The classic triad of hemolytic jaundice:
- Anemia — pallor, fatigue, exertional dyspnea, palpitations, tachycardia
- Jaundice (icterus) — lemon-yellow tint to skin and sclera (scleral icterus is the earliest sign, detectable when total bilirubin >2–3 mg/dL); typically mild to moderate (bilirubin rarely exceeds 4–5 mg/dL from hemolysis alone in a normally functioning liver)
- Splenomegaly — from RBC destruction + extramedullary hematopoiesis
Additional symptoms:
- Dark urine (excess urobilinogen → urobilin) — note: NOT bilirubinuria
- In intravascular hemolysis: port-wine/cola-colored urine (hemoglobinuria)
- Dark stools (excess stercobilin)
- Gallstone symptoms (RUQ pain, biliary colic) in chronic cases
- Frontal bossing and maxillary prominence (in thalassemia major, from marrow expansion)
- Leg ulcers (in sickle cell disease)
- Episodic painful crises (sickle cell)
- Fever, chills, back/flank pain during acute hemolytic episodes
7. Physical Examination
| Finding | Significance |
|---|
| Scleral icterus | First visible site of jaundice; best seen in natural daylight |
| Skin jaundice | Lemon-yellow hue (vs. orange-yellow in hepatic or green in obstructive) |
| Pallor (conjunctivae, palms, nailbeds) | Underlying anemia |
| Splenomegaly | Palpable on left side; may be massive in thalassemia, hereditary spherocytosis |
| Hepatomegaly | Mild; from Kupffer cell hyperplasia and hematopoiesis |
| Tachycardia, flow murmur | Compensatory hyperdynamic circulation from anemia |
| Frontal bossing, prominent malar eminences | Medullary expansion in thalassemia |
| Leg ulcers (medial malleolus) | Vaso-occlusion in sickle cell disease |
| Lymphadenopathy | In AIHA secondary to lymphoma/CLL |
| No signs of hepatic failure | (Distinguishes from hepatic/obstructive jaundice) — no spider angiomata, no asterixis, no ascites unless splenoportal hypertension |
| Pigment gallstones (Murphy's sign) | Biliary colic if stones formed |
8. Diagnostic Workup
Tier 1 — Initial Tests
| Test | Expected Finding in Hemolytic Jaundice |
|---|
| Total serum bilirubin | Elevated (often 2–5 mg/dL) |
| Direct (conjugated) bilirubin | Normal or slightly elevated (<15% of total) |
| Indirect (unconjugated) bilirubin | Markedly elevated (predominant fraction) |
| ALT / AST | Normal (distinguishes from hepatocellular disease) |
| Alkaline phosphatase | Normal (distinguishes from cholestasis) |
| CBC | Normocytic (or macrocytic with high reticulocytes) anemia; leukocytosis possible in crisis |
| Reticulocyte count | Elevated (>2%; often >5–10%) — compensatory erythropoiesis |
| Peripheral blood smear | Spherocytes (HS, AIHA), schistocytes (MAHA/TTP/HUS), sickle cells, target cells (thalassemia), Heinz bodies (G6PD after supravital stain) |
Tier 2 — Hemolysis Confirmation
| Test | Finding |
|---|
| Serum LDH | Elevated (released from lysed RBCs) |
| Serum haptoglobin | Decreased or absent (consumed binding free Hgb) |
| Serum free hemoglobin | Elevated (especially in intravascular) |
| Urine urobilinogen | Increased |
| Urine bilirubin (dipstick) | Absent (negative for bilirubinuria — key differentiator) |
| Urine hemoglobin / hemosiderinuria | Present in intravascular hemolysis |
| Fecal stercobilin | Increased |
Tier 3 — Specific Etiology
| Test | Diagnoses |
|---|
| Direct Coombs test (DAT) | Positive: AIHA, HDN, drug-induced; Negative: hereditary causes |
| G6PD enzyme assay | G6PD deficiency (note: may be falsely normal during/after acute crisis due to older G6PD-deficient cells being selectively destroyed) |
| Osmotic fragility test | Increased in hereditary spherocytosis |
| Hemoglobin electrophoresis | HbS (sickle cell), HbH (alpha-thalassemia), HbA2 elevated (beta-thalassemia) |
| Flow cytometry (CD55/CD59) | Absent in PNH |
| Bone marrow aspirate | Erythroid hyperplasia (M:E ratio decreased) |
| LDH isoforms / ADAMTS13 | ADAMTS13 deficiency in TTP |
| Cold agglutinin titer | Cold agglutinin disease |
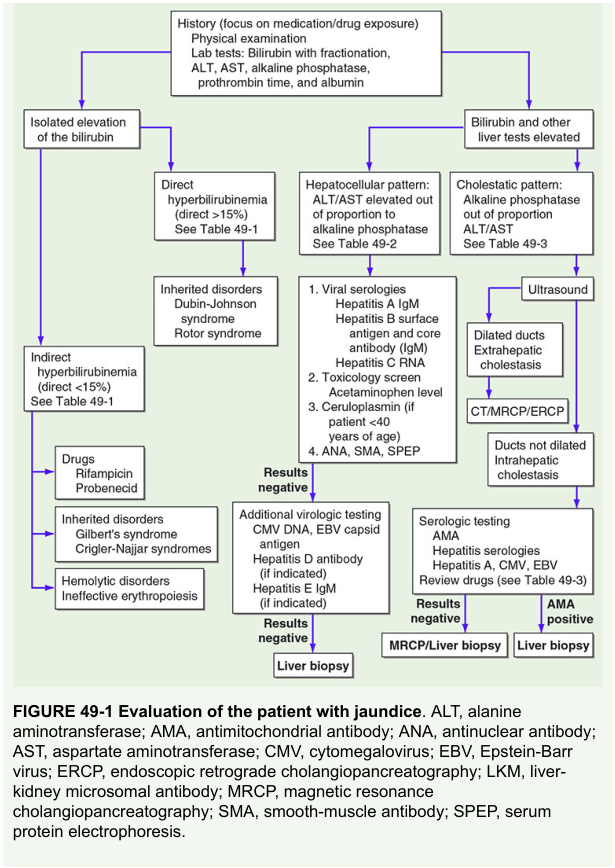
The diagnostic algorithm above (Harrison's Fig. 49-1) shows how isolated indirect hyperbilirubinemia points toward hemolytic disorders, inherited disorders (Gilbert's, Crigler-Najjar), or drug effects.
Key differentiating lab pattern:
| Parameter | Hemolytic | Hepatocellular | Obstructive |
|---|
| Indirect bilirubin | ↑↑ | ↑ | Normal/↑ |
| Direct bilirubin | Normal | ↑ | ↑↑ |
| ALT/AST | Normal | ↑↑ | Normal/↑ |
| ALP | Normal | ↑ | ↑↑ |
| Urine bilirubin | Absent | Present | Present |
| Urine urobilinogen | ↑↑ | ↑ | Absent |
| Haptoglobin | ↓ | Normal | Normal |
| LDH | ↑ | ↑ | Normal |
— Guyton and Hall, p. 864; Henry's Clinical Diagnosis and Management by Laboratory Methods; Lippincott Biochemistry, p. 800–803; Harrison's Principles of Internal Medicine, 22nd ed
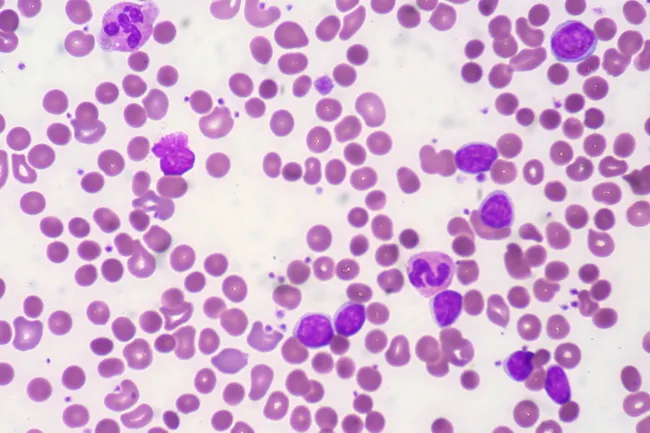
9. Peripheral Blood Smear Images
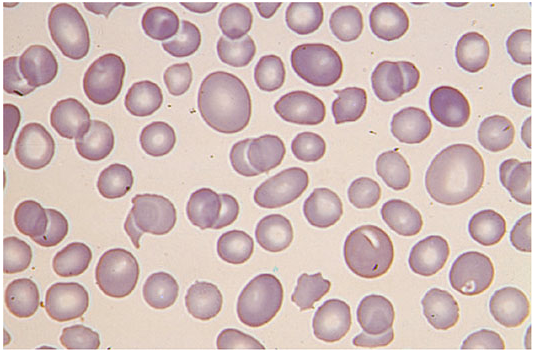
Spherocytes in hereditary spherocytosis — cells are small, dense, with no central pallor due to membrane loss. — Harrison's Principles of Internal Medicine, 21st ed, p. 1722
Schistocytes (fragmented RBCs, helmet cells) — hallmark of intravascular mechanical hemolysis in TTP/HUS/DIC.
AIHA in CLL — spherocytes with reduced central pallor, polychromasia, and elevated reticulocyte response.
10. Medications and Therapy
Treatment is etiology-specific:
Universal Support
- Folic acid (folate) 1–5 mg/day — all patients with chronic hemolysis; repletes stores depleted by accelerated erythropoiesis
- Blood transfusion — for acute severe anemia (Hgb < 7 g/dL or symptomatic); use washed/irradiated packed RBCs as appropriate
Disease-Specific
| Condition | Treatment |
|---|
| AIHA (warm-type) | Prednisone 1 mg/kg/day (first-line); Rituximab (anti-CD20) for steroid-refractory; Splenectomy; Immunosuppressants (azathioprine, mycophenolate) |
| Cold agglutinin disease | Rituximab; avoid cold exposure; Sutimlimab (anti-C1s complement inhibitor) |
| G6PD deficiency | Avoid triggers (oxidant drugs, fava beans, infections); Supportive care; Transfusion if severe |
| Hereditary spherocytosis | Splenectomy (curative for hemolysis; requires pre-op vaccination — pneumococcal, meningococcal, Hib); Cholecystectomy if gallstones |
| Sickle cell disease | Hydroxyurea (increases HbF → reduces sickling); Voxelotor (prevents Hgb polymerization); L-glutamine; Crizanlizumab; Chronic transfusion program; Hematopoietic stem cell transplant (curative) |
| Thalassemia major | Regular transfusions every 2–4 weeks; Deferasirox / deferoxamine (iron chelation); Luspatercept; HSCT (curative) |
| PNH | Eculizumab or ravulizumab (anti-C5 complement inhibitor — highly effective); HSCT in aplastic/thrombotic complications |
| TTP | Therapeutic plasma exchange (TPE — STAT); Caplacizumab; Steroids; Rituximab |
| HUS (typical/Stx-mediated) | Supportive; Eculizumab for atypical HUS |
| Neonatal hemolytic disease / Kernicterus risk | Phototherapy (converts UCB to water-soluble isomers); Exchange transfusion if bilirubin critically elevated |
| Malaria | Antimalarials (chloroquine, artemisinin-based combination therapy, quinine) |
| Post-splenectomy | Penicillin prophylaxis; Vaccines (every 5 years for pneumococcal) |
11. Related Syndromes
Inherited Disorders of Bilirubin Metabolism (Differential Diagnosis)
| Syndrome | Defect | Bilirubin Type | Features |
|---|
| Gilbert syndrome | Mild ↓ UGT1A1 activity (~30%); also ↓ OATP uptake | Mild unconjugated ↑ (<3 mg/dL) | Benign; exacerbated by fasting, stress, illness; no hemolysis needed |
| Crigler-Najjar Type I | Complete absence of UGT1A1 | Severe unconjugated ↑ (>20 mg/dL) | Kernicterus in infancy; requires ~12 hrs/day phototherapy; fatal without liver transplant |
| Crigler-Najjar Type II (Arias) | Severe ↓ UGT1A1 (residual activity) | Unconjugated ↑ (6–20 mg/dL) | Less severe; responds to phenobarbital (induces UGT1A1) |
| Dubin-Johnson syndrome | MRP2 (ABCC2) defect → ↓ CB secretion into bile | Conjugated ↑ | Benign; liver appears black/darkly pigmented; urine coproporphyrin I:III ratio reversed |
| Rotor syndrome | OATP1B1/1B3 defect | Conjugated ↑ | No liver pigmentation; similar presentation to DJS |
Other Related Syndromes
- Hereditary spherocytosis — most common inherited hemolytic anemia in Northern Europeans; autosomal dominant (mostly); gene mutations: ANK1, SPTB, SPTA1, EPB42, SLC4A1
- Hereditary elliptocytosis — EPB41, SPTA1 mutations
- G6PD deficiency — X-linked; most common RBC enzyme defect worldwide; Mediterranean, African, Asian variants
- Sickle cell disease — homozygous HbS; autosomal recessive
- Beta-thalassemia — ↓/absent beta-globin chains; autosomal recessive
- Hemolytic disease of the newborn (HDN / erythroblastosis fetalis) — Rh or ABO incompatibility; maternal IgG crosses placenta
- TTP (Thrombotic Thrombocytopenic Purpura) — ADAMTS13 deficiency → ultra-large vWF multimers → platelet thrombi → MAHA + thrombocytopenia + fever + neurological symptoms + renal failure (pentad)
- HUS (Hemolytic Uremic Syndrome) — triad: MAHA + thrombocytopenia + acute kidney injury; typical form from Shiga toxin (E. coli O157:H7)
- Blackwater fever — severe intravascular hemolysis in malaria (P. falciparum), causing hemoglobinuria → dark urine
12. Complications
| Complication | Mechanism |
|---|
| Pigment (bilirubin) gallstones | Chronic excess bilirubin → precipitation of unconjugated bilirubin calcium salts; cholecystitis, biliary colic, obstructive jaundice |
| Aplastic crisis | Parvovirus B19 infects erythroid precursors → sudden cessation of RBC production → life-threatening anemia (Hgb may fall precipitously) |
| Hemolytic crisis | Acute exacerbation of hemolysis (infection, oxidant exposure) → severe anemia, jaundice, hemoglobinuria |
| Megaloblastic crisis | Folate deficiency from chronic high RBC turnover → failed erythropoiesis → pancytopenia |
| Kernicterus | UCB (especially in neonates) crosses BBB → deposits in basal ganglia and brain → neurological damage (choreoathetosis, hearing loss, intellectual disability, death) |
| Pulmonary hypertension | Chronic hemolysis → NO scavenging by free Hgb → vasoconstriction; especially in sickle cell and PNH |
| Iron overload (hemosiderosis) | In chronically transfused patients (thalassemia, SCD) → cardiac, hepatic, endocrine damage; prevented with chelation |
| Splenic sequestration crisis | Acute massive pooling of RBCs in spleen (especially SCD in children) → sudden severe anemia |
| Post-splenectomy sepsis | Overwhelming infection with encapsulated bacteria (S. pneumoniae, H. influenzae, N. meningitidis); prevented by vaccines + prophylactic penicillin |
| Chronic kidney disease | Chronic hemoglobinuria and tubular hemosiderin deposition (especially in PNH) |
| Venous thromboembolism | PNH, sickle cell → hypercoagulability |
| Growth retardation / skeletal deformities | Chronic hemolysis in childhood (thalassemia, SCD) → marrow expansion |
Summary Table
| Feature | Hemolytic Jaundice |
|---|
| Type | Pre-hepatic (overproduction) |
| Bilirubin | ↑ Unconjugated (indirect); direct normal |
| Urine bilirubin | Absent |
| Urine urobilinogen | ↑↑ |
| Stool color | Dark (excess stercobilin) |
| Haptoglobin | ↓ or absent |
| LDH | ↑ |
| Reticulocytes | ↑ |
| Liver enzymes (ALT/AST/ALP) | Normal |
| Smear | Spherocytes, schistocytes, sickle cells, etc. |
| Coombs test | Positive (AIHA); Negative (hereditary) |
| Classic triad | Anemia + Jaundice + Splenomegaly |
Sources:
- Biochemistry, 8th ed, Lippincott Illustrated Reviews — pp. 794–803
- Guyton and Hall Textbook of Medical Physiology — pp. 862–864
- Harrison's Principles of Internal Medicine, 21st & 22nd Editions
- Harper's Illustrated Biochemistry, 32nd ed
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Robbins & Kumar Pathologic Basis of Disease
- Rosen's Emergency Medicine