Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the material needed for a comprehensive answer. Here is a structured explanation of the mechanism of ARDS.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
Definition and Core Concept
ARDS is a syndrome of non-cardiogenic pulmonary edema caused by increased alveolar-capillary permeability rather than elevated hydrostatic pressure (as in heart failure). The edema fluid is protein-rich and exudative, flooding the alveoli and causing profound hypoxemia and decreased lung compliance. — Murray & Nadel's Textbook of Respiratory Medicine
Triggers and Risk Factors
ARDS is initiated by either direct (pulmonary) or indirect (non-pulmonary) injury:
| Direct Injury (Pulmonary) | Indirect Injury (Non-pulmonary) |
|---|---|
| Pneumonia (bacterial, viral, COVID-19) | Sepsis |
| Aspiration of gastric contents | Major trauma |
| Pulmonary contusion | Multiple blood transfusions |
| Toxic inhalation | Pancreatitis |
| Near-drowning | Cardiopulmonary bypass |
| Reperfusion injury | Drug overdose |
Sepsis, trauma, aspiration, and massive transfusions carry the highest risk. Presence of more than one risk factor further increases incidence.
Pathological Phases: Diffuse Alveolar Damage (DAD)
The pathologic hallmark of ARDS is diffuse alveolar damage (DAD), which progresses through three overlapping stages:
1. Exudative Phase (Days 1–7)
- Protein-rich fluid and hyaline membranes (composed of cellular debris, proteins, and surfactant components) fill alveolar spaces
- Widespread epithelial disruption
- Intense neutrophil infiltration of the interstitium and airspaces
- Impaired gas exchange → profound hypoxemia
2. Proliferative Phase (~Day 7 onwards)
- Hyaline membranes are reorganized
- Early fibrosis appears
- Obliteration of pulmonary capillaries
- Deposition of interstitial and alveolar collagen
- Reduction in neutrophil numbers
3. Fibrotic Phase (>2 weeks)
- Pulmonary fibrosis in a subset of patients with persistent ARDS
- Elevated N-terminal procollagen peptide III in BAL fluid (detectable as early as 24 hours after onset) suggests fibroproliferation begins simultaneously with — not after — inflammatory injury
Cellular and Molecular Mechanisms
Step 1: Alveolar-Capillary Barrier Breakdown
Damage to the alveolar epithelium is considered the key precipitating event. Multiple mechanisms contribute to epithelial cell death:
- Direct cytotoxicity
- Apoptosis
- Necrosis triggered by inflammatory mediators
The microvascular endothelium is also damaged, and together these two layers form a compromised barrier that allows protein-rich fluid to flood the alveoli.
Step 2: Neutrophil Sequestration and Activation
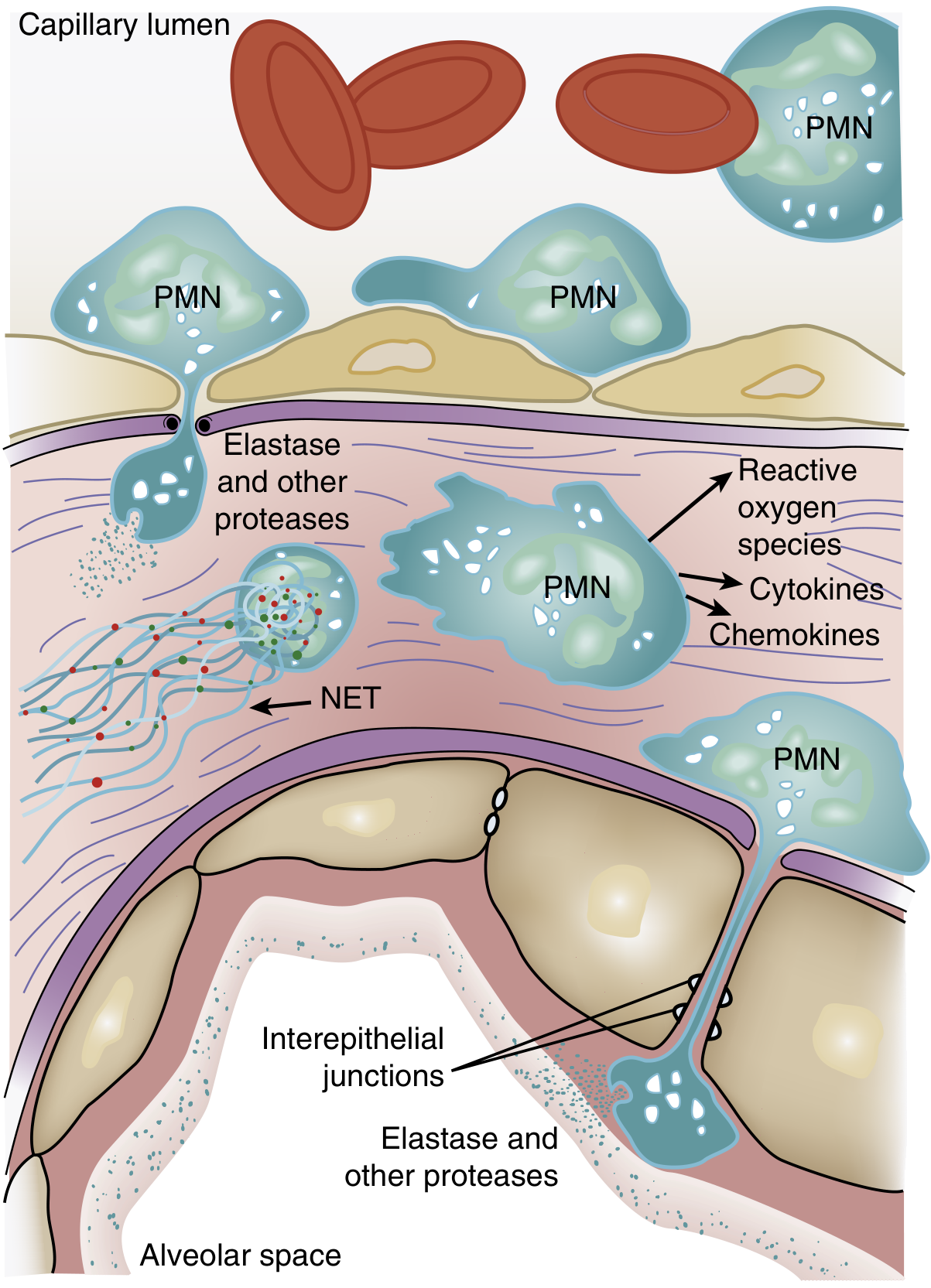
Figure: Role of neutrophils in ARDS. PMNs transmigrate from the capillary lumen through the endothelial and epithelial barriers into the alveolar space, releasing destructive mediators at each step. — Murray & Nadel's Textbook of Respiratory Medicine
One of the earliest manifestations of ARDS — even before hypoxemia — is a transient leukopenia from neutrophil sequestration in the pulmonary microvasculature:
- Pulmonary capillaries are narrower than neutrophils; passage requires cell deformation
- Activated neutrophils become "stiff" (due to actin cytoskeletal changes) and become trapped
- Sequestered neutrophils then migrate into lung parenchyma, disrupting endothelial barrier integrity
Once in the interstitium and alveoli, activated neutrophils release:
- Reactive oxygen species (ROS) — oxidative injury to cells
- Proteolytic enzymes (e.g., leukocyte elastase) — digest structural proteins
- Cationic peptides (e.g., defensins)
- Eicosanoids
- TNF-α and IL-1β — amplify the inflammatory cascade
- Neutrophil extracellular traps (NETs) — chromatin fibers that trap pathogens but also damage host tissue
Step 3: Cytokine Storm and Amplification
The inflammatory response is amplified by a network of cytokines and chemokines:
- TNF-α and IL-1β upregulate adhesion molecules, recruit more neutrophils, and promote vascular permeability
- Macrophages and epithelial cells further release pro-inflammatory mediators
- The transcription factor NF-κB is a master regulator of this inflammatory amplification
- Heat shock protein 70 (HSP70) normally suppresses NF-κB; loss of HSP70 is associated with increased lung injury
Step 4: Surfactant Dysfunction
Injured type II pneumocytes produce abnormal surfactant:
- Surfactant composition is altered
- Surfactant proteins (SP-A, SP-B, SP-C, SP-D) are reduced
- Phospholipase A2 (released in pancreatitis-associated ARDS) enzymatically degrades surfactant
- Loss of surfactant → alveolar collapse → worsened V/Q mismatch and shunting
Step 5: Impaired Alveolar Fluid Clearance
Normally, Na⁺ channels on type II pneumocytes drive fluid reabsorption from alveoli. In ARDS:
- Hypoxia impairs expression of epithelial Na⁺ channel subunits
- Hypoxia also directly inhibits apical Na⁺ channel activity and basolateral Na⁺/K⁺-ATPase
- Increased nitric oxide (e.g., from hemorrhagic shock) impairs β-adrenergic–mediated fluid clearance
- Result: edema fluid accumulates and cannot be cleared
Step 6: Endothelial Dysfunction — Angiopoietin Imbalance
Angiopoietin 2 (Ang2), released by injured endothelial cells, promotes vascular leak:
- Elevated Ang2 is found in patients with sepsis and ARDS
- Ang2 competitively antagonizes Angiopoietin 1 (Ang1), which normally maintains endothelial barrier integrity via the Tie2 receptor
- Genetic variants in Ang2 are associated with increased ARDS risk
- This imbalance further compromises endothelial barrier function
Physiological Consequences
| Consequence | Mechanism |
|---|---|
| Profound hypoxemia | Alveolar flooding → intrapulmonary right-to-left shunt + low V/Q regions |
| Decreased compliance | Alveolar edema, atelectasis, surfactant loss |
| Increased dead space | Vascular obliteration → elevated minute ventilation requirement |
| Pulmonary hypertension | Hypoxic vasoconstriction, intravascular fibrin deposition, compression by positive-pressure ventilation |
| Hypercapnia (in severe cases) | Massively increased dead space overwhelms compensatory hyperventilation |
Summary Cascade
Trigger (sepsis, pneumonia, trauma, etc.)
↓
Direct or indirect lung injury
↓
Alveolar epithelial + endothelial damage
↓
Neutrophil sequestration → transmigration
↓
Release of ROS, proteases, NETs, cytokines (TNF-α, IL-1β)
↓
Increased alveolar-capillary permeability
↓
Protein-rich exudate floods alveoli → Hyaline membrane formation (DAD)
↓
Surfactant dysfunction + impaired Na⁺-driven fluid clearance
↓
Alveolar collapse, shunt, hypoxemia, ↓ compliance, pulmonary hypertension
↓
ARDS
Sources: Murray & Nadel's Textbook of Respiratory Medicine; Goldman-Cecil Medicine; Sabiston Textbook of Surgery
This is a shared conversation. Sign in to Orris to start your own chat.