Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have rich, comprehensive content from Murray & Nadel's Textbook of Respiratory Medicine to construct a thorough mechanistic explanation. Let me synthesize everything.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
Definition and Core Pathophysiology
ARDS is a syndrome of noncardiogenic pulmonary edema caused by increased alveolar-capillary permeability rather than elevated hydrostatic pressure (as in heart failure). The edema fluid is protein-rich and exudative, filling alveoli and causing:
- Severely decreased respiratory system compliance
- Profound refractory hypoxemia (right-to-left shunting)
- Markedly increased dead space ventilation
- Pulmonary hypertension
— Murray & Nadel's Textbook of Respiratory Medicine, block31
Phases of ARDS (Diffuse Alveolar Damage)
The pathological correlate of ARDS is diffuse alveolar damage (DAD), progressing through three overlapping stages:
1. Exudative Phase (Days 0–7)
- Hyaline membranes (composed of cellular debris, fibrin, proteins, and surfactant components) line the denuded alveolar walls
- Protein-rich fluid floods the airspaces
- Widespread epithelial disruption — both type I and type II pneumocytes are injured
- Massive neutrophil infiltration of the interstitium and airspaces
2. Proliferative Phase (~Days 7–14)
- Hyaline membranes are reabsorbed and reorganized
- Fibrosis begins to appear
- Pulmonary capillary obliteration and interstitial/alveolar collagen deposition
- Neutrophil numbers decrease; alveolar macrophages increase
3. Fibrotic Phase (>2 weeks, in some patients)
- Progressive pulmonary fibrosis in a subset of patients
- Collagen synthesis (detected via N-terminal procollagen peptide III in BAL fluid) can begin as early as 24 hours, suggesting fibroproliferation may start simultaneously with inflammatory injury rather than after it
— Murray & Nadel's Textbook of Respiratory Medicine, block31
The Central Lesion: Alveolar-Capillary Barrier Disruption
The alveolar-capillary barrier has two components, both of which are injured:
Endothelial Injury
- The pulmonary microvascular endothelium is disrupted, increasing vascular permeability
- Fluid leaks from capillaries into the interstitium and then into alveolar spaces
- Plasma proteins, including clotting factors, enter the alveolus — contributing to hyaline membrane formation and fibrin deposition
Epithelial Injury
- Type I pneumocytes (covering ~95% of the alveolar surface) are highly vulnerable and are destroyed early
- Loss of type I cells removes the barrier that keeps alveoli dry and allows massive alveolar flooding
- Type II pneumocytes (cuboidal, comprising the remaining ~5%) are more resistant but are also injured; they are critical because they:
- Produce surfactant — loss leads to alveolar collapse and worsened hypoxemia
- Are the progenitor cells that regenerate type I pneumocytes during repair
- Injury to type II cells also impairs active sodium transport (via Na⁺/K⁺-ATPase and epithelial sodium channels), which normally drives alveolar fluid clearance — the mechanism by which alveolar flooding is cleared
— Murray & Nadel's Textbook of Respiratory Medicine, block31
Cellular Mechanisms of Injury
Neutrophils — The Primary Effector Cells
Neutrophil accumulation in the alveolus and pulmonary vasculature is a hallmark of ARDS:
- Triggering cytokines: Systemic triggers (sepsis, trauma, aspiration) activate macrophages and the complement system, releasing IL-8, IL-1β, TNF-α, and C5a that recruit neutrophils to the lung
- Neutrophil adherence: Upregulation of endothelial adhesion molecules (ICAM-1, E-selectin, VCAM-1 via NF-κB signaling) promotes neutrophil margination and transmigration
- Oxidants: Activated neutrophils generate reactive oxygen species (ROS) via NADPH oxidase, causing direct membrane lipid peroxidation and protein oxidation
- Proteases: Neutrophil elastase (NE) and metalloproteinases degrade structural proteins; NE specifically cleaves epithelial and endothelial cadherins — the proteins holding adherens junctions together — precipitating cell detachment and alveolar flooding
- Neutrophil extracellular traps (NETs): Web-like structures of DNA, histones, and antimicrobial peptides (myeloperoxidase, NE, cathepsin G) that are released en masse in sepsis. NETs cause endothelial damage and thrombus formation, and in animal models of ARDS, NET formation correlated with severe lung structural destruction; DNase treatment attenuated injury
- Phosphatidylinositol 3-kinase-γ (PI3Kγ): A key signaling node in neutrophils activated by IL-8 or fMLP, governing neutrophil recruitment and cytokine production
Importantly, ARDS can occur in profoundly neutropenic patients, indicating that neutrophils are not strictly required. In neutropenic ARDS, alveolar macrophages may be the primary source of injury.
— Murray & Nadel's Textbook of Respiratory Medicine, block31
Alveolar Macrophages
- Resident alveolar macrophages are the sentinels of the lung, recognizing DAMPs (danger-associated molecular patterns) and PAMPs (pathogen-associated molecular patterns) via Toll-like receptors
- Upon activation, they secrete TNF-α, IL-1β, IL-6, IL-8, and platelet-activating factor (PAF), amplifying neutrophil recruitment and systemic inflammation
Coagulation Dysregulation
- The injured alveolus generates a procoagulant microenvironment: plasma proteins (including fibrinogen and clotting factors) flood the airspace and fibrin is deposited → hyaline membranes
- Intravascular fibrin deposition in pulmonary vessels contributes to pulmonary hypertension
- Impaired fibrinolysis compounds this — BAL fluid in ARDS shows elevated plasminogen activator inhibitor-1 (PAI-1) and depressed tissue plasminogen activator (tPA) activity
Surfactant Dysfunction
- Surfactant (produced by type II pneumocytes) reduces surface tension at the air-liquid interface and prevents alveolar collapse at end-expiration
- In ARDS, surfactant is depleted by:
- Type II pneumocyte injury/death
- Dilution and inactivation by protein-rich edema fluid (plasma proteins competitively displace surfactant from the alveolar surface)
- Phospholipase A₂ (released during pancreatitis and by activated inflammatory cells) enzymatically degrades surfactant phospholipids
- The result is alveolar collapse, further impairment of oxygenation, and reduced lung compliance
Pulmonary Hypertension in ARDS
Multiple mechanisms converge:
- Hypoxic pulmonary vasoconstriction (HPV) — the normal response to alveolar hypoxia causes vasoconstriction, diverting blood from flooded units, but at the cost of elevated pulmonary arterial pressure
- Intravascular fibrin deposition — thrombi in small pulmonary vessels increase resistance
- Compression of blood vessels by positive-pressure mechanical ventilation
- Vascular remodeling in the fibrotic phase
Ventilator-Induced Lung Injury (VILI) and Biotrauma
Once a patient with ARDS is placed on mechanical ventilation, the ventilator itself can perpetuate and amplify injury:
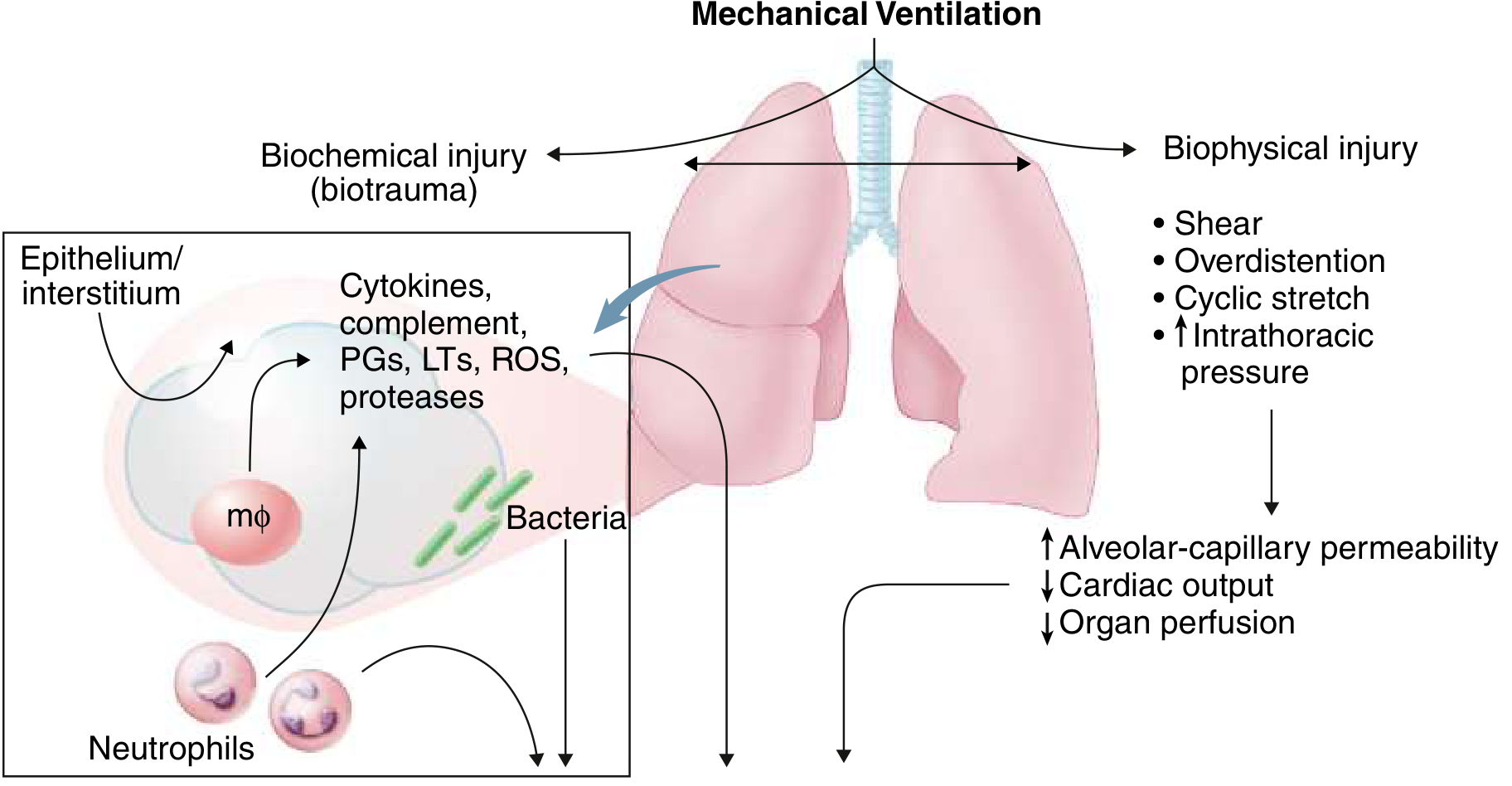
- Biophysical injury: Alveolar overdistension (volutrauma), cyclic shear stress from opening and closing of unstable alveoli (atelectrauma), and elevated intrathoracic pressure
- Biotrauma: Mechanical stretch of the lung epithelium stimulates release of cytokines (TNF-α, IL-1β, IL-6, IL-8), prostaglandins, leukotrienes, ROS, and proteases into the systemic circulation — a likely driver of multi-organ failure
- This explains why most patients with fatal ARDS die of sepsis and multi-organ failure, not refractory hypoxemia
— Goldman-Cecil Medicine, block12; Murray & Nadel's block31
Impaired Alveolar Fluid Clearance
Normally, type II pneumocytes actively transport sodium from the alveolar lumen into the interstitium via apical epithelial sodium channels (ENaC) and basolateral Na⁺/K⁺-ATPase, with water following osmotically. In ARDS:
- Type II cell injury disrupts this transport mechanism
- Alveolar flooding therefore cannot be cleared efficiently
- Catecholamines and β-agonists can upregulate ENaC and Na⁺/K⁺-ATPase activity, which is part of the rationale for β-agonist trials in ARDS (though clinical benefit has not been conclusively demonstrated)
Summary Cascade
Triggering insult (direct: pneumonia, aspiration | indirect: sepsis, trauma, pancreatitis)
↓
Macrophage/complement activation → IL-1β, TNF-α, IL-8, C5a
↓
Neutrophil recruitment to lung (ICAM-1 ↑, E-selectin ↑)
↓
Neutrophil activation → ROS, elastase, NETs, metalloproteinases
↓
Endothelial + Epithelial barrier disruption
↓
Protein-rich exudate floods alveoli → hyaline membranes (DAD)
↓
Surfactant loss → alveolar collapse → ↓ compliance, V/Q mismatch, shunt → hypoxemia
↓
Pulmonary hypertension (HPV + fibrin deposition)
↓
Mechanical ventilation → VILI/biotrauma → multi-organ failure
Sources: Murray & Nadel's Textbook of Respiratory Medicine; Goldman-Cecil Medicine International Edition
This is a shared conversation. Sign in to Orris to start your own chat.