Neurolathyrism community medicine k park
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have comprehensive information. K. Park's textbook is not in the library, but the content from standard neurological textbooks covers what K. Park discusses on neurolathyrism from a community medicine perspective. Let me compile a thorough response.
Neurolathyrism
Definition
Neurolathyrism is a chronic toxic nutritional neurological disease caused by prolonged or subacute ingestion of flour made from the drought-resistant grass pea (Lathyrus sativus), also known as chickling vetch, vetch pea, or khesari dal. It is an important example of a selective upper motor neuron (UMN) disorder caused by a natural food-borne excitotoxin.
Causative Agent / Neurotoxin
The responsible neurotoxin is β-N-oxalylamino-L-alanine (BOAA) — an AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid) glutamate receptor agonist.
Mechanism:
- BOAA over-stimulates AMPA glutamate receptors
- Leads to increased intracellular reactive oxygen species (ROS)
- Impairs mitochondrial oxidative phosphorylation
- Causes selective degeneration of Betz cells in the motor cortex and the longest corticospinal tracts subserving lower-limb function
- Anterior horn cells are spared
Epidemiology (Community Medicine Perspective)
| Feature | Details |
|---|---|
| Geographic distribution | India, Bangladesh, Ethiopia, China, Romania, Spain; epidemic in regional concentration camps during WWII |
| Occurrence | Endemic in drought-prone areas; can become epidemic during famine/food shortage |
| High-risk group | Malnourished populations; one Ethiopian epidemic showed higher incidence in boys aged 10–14 years |
| Risk factor | Cooking grass pea foods in traditional clay pots (increases toxin availability) |
| Trigger | Excess consumption of L. sativus flour when wheat/other grains are scarce |
| Historical note | Known since ancient times — mentioned by Hippocrates, Pliny, Galen, and Avicenna |
Clinical Features
Onset: Acute or chronic; may follow months of near-exclusive L. sativus consumption
Presenting symptoms:
- Pain, paresthesias (formications/tingling), and weakness in the lower extremities
- Progressive spastic paraplegia (bilateral)
- Leg cramps and spasms
- Bladder dysfunction — frequency, urgency, sphincteric spasms
- Erectile dysfunction
- Occasional coarse tremor of upper extremities
Key characteristics:
- Predominantly upper motor neuron signs (spasticity, hyperreflexia, extensor plantar response)
- Sensation is usually preserved (though some historical cases reported sensory loss)
- Once established, the disorder is irreversible but non-progressive (unless exposure continues)
- Lifespan is not affected
Neuropathology
- Degeneration of corticospinal tracts and posterior columns in the spinal cord
- Loss of myelinated fibers in lateral and posterior columns
- Loss of large Betz cells in motor cortex
- Anterior horn cells are unaffected (distinguishing from ALS/polio)
- Gliosis and thickening of blood vessel walls in degenerated tracts
Prevention (Community Medicine Focus)
| Measure | Details |
|---|---|
| Primary | Avoid excessive/exclusive consumption of L. sativus |
| Detoxification | Soaking, boiling, and discarding water from grass peas reduces BOAA content |
| Dietary diversification | Ensure adequate protein and varied diet, especially during famines |
| Public health measures | Education, food security programs, supply of alternative grains during drought |
| Pot change | Avoid cooking in clay pots (which may increase toxin bioavailability) |
Differential Diagnosis
- Konzo (cyanide toxicity from cassava — similar UMN picture)
- Tropical spastic paraparesis (HTLV-I)
- Hereditary spastic paraplegia
- Primary lateral sclerosis
- Spinal cord compression
Key Points for Exams
- Cause: BOAA toxin in Lathyrus sativus (grass pea / khesari dal)
- Mechanism: Excitotoxicity via AMPA receptor over-activation
- Pathology: Upper motor neuron disease; Betz cells degenerate; anterior horn cells spared
- Features: Irreversible spastic paraplegia, no sensory loss typically, non-progressive
- Occurs during famines/droughts — a classic community medicine/public health topic
- Prevention: Dietary diversification, detoxification of peas before consumption
Sources: Adams and Victor's Principles of Neurology, 12th Ed. | Bradley and Daroff's Neurology in Clinical Practice | Harper's Illustrated Biochemistry, 32nd Ed.
Natural history of diseases k park community medicine
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
K. Park's "Textbook of Preventive and Social Medicine" is not in the library database. However, this is a core community medicine topic that I know thoroughly from the standard curriculum. I'll provide a comprehensive answer based on the established content of K. Park, which is well-defined in the public health literature.
Natural History of Disease
(As per K. Park — Preventive & Social Medicine)
Definition
The natural history of disease refers to the way in which a disease process evolves over time — from its earliest biological beginnings to its final outcome — in the absence of medical intervention. Understanding it is fundamental to designing preventive strategies at each stage.
The concept was classically described by Leavell and Clark (1965).
Two Periods in Natural History of Disease
1. Pre-pathogenesis Period
The period before the disease process begins in the host. The disease agent exists in the environment and interacts with host and environmental factors, but no pathological changes have yet occurred in the host.
- The ecological triad (Agent – Host – Environment) is in equilibrium or being disturbed
- The host is susceptible but not yet diseased
- The appropriate intervention here is Primary Prevention
2. Pathogenesis Period
The period during which pathological changes occur in the host. Begins when the agent enters a susceptible host.
Divided into two stages:
a) Early Pathogenesis (Sub-clinical / Pre-symptomatic)
- Biological/pathological changes are occurring
- No signs or symptoms yet detectable clinically
- May be detectable by screening tests
- Appropriate intervention: Secondary Prevention (early diagnosis and treatment)
b) Late Pathogenesis (Clinical Disease)
- Symptoms and signs become manifest
- Can progress to:
- Recovery
- Disability
- Death
Leavell & Clark's Model: Levels of Prevention
| Level | Period | Interventions |
|---|---|---|
| Primary Prevention | Pre-pathogenesis | Health promotion + Specific protection |
| Secondary Prevention | Early pathogenesis | Early diagnosis + Prompt treatment |
| Tertiary Prevention | Late pathogenesis | Disability limitation + Rehabilitation |
Primary Prevention
Applied during the pre-pathogenesis period.
A. Health Promotion (non-specific, raises general resistance):
- Health education
- Adequate nutrition
- Provision of adequate housing, recreation, working conditions
- Marriage counselling and sex education
- Attention to personality development
- Genetic counselling
B. Specific Protection (targets specific agents/diseases):
- Immunisation against specific diseases
- Use of specific nutrients (e.g. iodine in salt for goitre)
- Protection from occupational hazards
- Protection from accidents
- Use of specific nutrients (vitamins)
- Environmental sanitation
- Protection from carcinogens
Secondary Prevention
Applied during early pathogenesis — aims to halt or reverse the disease process.
A. Early Diagnosis and Prompt Treatment:
- Case-finding surveys (mass/selective screening)
- Screening and follow-up
- Objectives: cure, prevent spread, prevent complications
B. Disability Limitation:
- Adequate treatment to prevent disability (applied in late pathogenesis)
- Provision of facilities to limit disability
Tertiary Prevention
Applied during late pathogenesis/clinical disease — minimises the effects of disease and disability.
A. Rehabilitation:
- Medical rehabilitation (maximum physical capacity)
- Social rehabilitation (restore social functioning)
- Vocational rehabilitation (gainful employment)
- Psychological rehabilitation
Concept of the "Iceberg of Disease"
A critical community medicine concept related to natural history:
/\ ← Clinical cases (visible above waterline)
/ \
/ \
───────────────── ← Waterline (clinical threshold)
/ \
/ Sub- \
/ clinical \ ← Undiagnosed / sub-clinical cases (below waterline)
/ Susceptibles \
/________________\ ← Susceptibles
- The tip of the iceberg = symptomatic/diagnosed cases (what clinicians see)
- The submerged portion = sub-clinical infections, undiagnosed cases, carriers, and susceptibles
- This "hidden" mass is the reservoir from which clinical cases emerge
- Screening programs aim to detect sub-clinical disease before it surfaces
Examples by iceberg pattern:
| Disease | Iceberg Characteristic |
|---|---|
| Polio | Large submerged part (most infections sub-clinical) |
| Tuberculosis | Large submerged part (latent TB >> active TB) |
| Diabetes / HTN | Large submerged portion (many undiagnosed) |
| Rabies | Small/no submerged part (nearly always clinical) |
Spectrum of Disease
The natural history produces a spectrum of outcomes ranging from:
Subclinical infection → Mild disease → Moderate disease → Severe disease → Death
Also called the "gradient of infection" — not all exposed individuals develop the same severity of illness. Factors determining position on the spectrum:
- Agent factors (virulence, pathogenicity, dose)
- Host factors (immunity, nutrition, genetics)
- Environmental factors
Significance in Community Medicine
- Identifies the right point for intervention at each stage
- Guides screening program design (targeting sub-clinical disease)
- Explains why prevention is better than cure — intervening early (pre-pathogenesis) is most effective
- Forms the basis of the Leavell & Clark triad of prevention
- Helps estimate true disease burden (iceberg concept)
- Informs the design of health education messages
Note: K. Park's "Textbook of Preventive and Social Medicine" is not available in this library's collection. This answer is compiled from the established content of that text as it appears in the standard community medicine curriculum, cross-referenced with principles from Harrison's, Goldman-Cecil, and Family Medicine textbooks available in the library.
Screening of diseases
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
I now have rich, authoritative content. Let me compile a comprehensive answer.
Screening of Diseases
(Community Medicine / Preventive Medicine — K. Park Framework)
Definition
Screening is the presumptive identification of unrecognised disease or defect by the application of tests, examinations, or other procedures that can be applied rapidly to distinguish apparently well persons who probably have a disease from those who probably do not.
A screening test is not diagnostic — it identifies suspects who require further definitive investigation.
— Harrison's Principles of Internal Medicine, 22nd Ed.
Purpose / Objectives
- Early detection of disease in its pre-symptomatic phase (secondary prevention)
- Reduce morbidity and mortality from disease
- Identify high-risk individuals for targeted intervention
- Estimate the prevalence of a condition in a community
Types of Screening
| Type | Description | Example |
|---|---|---|
| Mass screening | Entire population screened | Neonatal heel-prick tests |
| Selective (high-risk) screening | Targeted at defined high-risk group | Diabetic screening in obese individuals |
| Multiphasic screening | Multiple tests applied simultaneously | Annual health check-ups |
| Opportunistic screening | During a routine clinical visit | BP measurement at a GP visit |
| Case-finding | Clinician screens patients already attending | Thyroid testing in women >50 |
Wilson & Jungner Criteria (WHO, 1968)
"Principles of Screening"
These are the gold-standard criteria for deciding whether a disease is suitable for screening:
- The condition should be an important health problem
- There should be an accepted treatment for patients with the disease
- Facilities for diagnosis and treatment should be available
- There should be a recognisable latent or early symptomatic stage
- There should be a suitable test or examination for the disease
- The test should be acceptable to the population
- The natural history of the disease should be adequately understood
- There should be an agreed policy on whom to treat
- The cost of case-finding (including diagnosis and treatment) should be economically balanced in relation to possible expenditure on medical care as a whole
- Case-finding should be a continuous process and not a once-and-for-all project
Validity of a Screening Test
The accuracy of a screening test is measured by four indices. Using the standard 2×2 table:
Disease PRESENT Disease ABSENT
Test POSITIVE a (TP) b (FP)
Test NEGATIVE c (FN) d (TN)
| Measure | Formula | Meaning |
|---|---|---|
| Sensitivity | a / (a+c) | Ability to detect disease when present (true positive rate) |
| Specificity | d / (b+d) | Ability to exclude disease when absent (true negative rate) |
| PPV (Positive Predictive Value) | a / (a+b) | Proportion of positive tests that are true positives |
| NPV (Negative Predictive Value) | d / (c+d) | Proportion of negative tests that are true negatives |
Key relationships:
- Sensitivity and specificity are relatively independent of prevalence
- PPV and NPV are strongly dependent on prevalence (disease frequency in the population)
- High prevalence → higher PPV for the same sensitivity/specificity
- Screening is most efficient when disease prevalence is high in the target population
Trade-off: Sensitivity vs Specificity
- Lowering the cut-off → ↑ Sensitivity, ↓ Specificity (more FP, fewer FN)
- Raising the cut-off → ↑ Specificity, ↓ Sensitivity (fewer FP, more FN)
- Ideal test: High sensitivity + High specificity
Reliability (Reproducibility) of a Screening Test
Reliability = the ability of a test to give consistent results on repeated application under the same conditions.
Factors affecting reliability:
- Observer variation (intra-observer and inter-observer)
- Biological variation in the subject
- Test methodology/equipment variation
Measured by Kappa statistic — degree of agreement beyond chance.
Yield of Screening
The number of cases detected by a screening programme. Determined by:
- Prevalence of unrecognised disease
- Sensitivity of the test
- The proportion of the population that agrees to be screened
- Number of previously screened individuals in the group
Biases in Screening
These biases can make screening appear beneficial even when it is not:
| Bias | Description |
|---|---|
| Lead-time bias | Screen detects disease earlier, but survival time is measured from diagnosis — survival appears longer without true benefit |
| Length-biased sampling | Screening preferentially detects slow-growing, less aggressive disease (which has a longer detectable pre-clinical phase), giving false impression of better prognosis |
| Overdiagnosis bias | Detecting disease that would never have caused symptoms/death in the patient's lifetime (e.g., indolent prostate cancer) |
| Selection bias | "Healthy screener effect" — those who attend screening are often healthier than average, inflating apparent benefit |
Harms of Screening
- False-positive results → unnecessary anxiety, further invasive tests
- False-negative results → false reassurance, delayed diagnosis
- Overdiagnosis → unnecessary treatment with attendant side effects
- Radiation exposure (mammography, CT)
- Psychological harm (anxiety during the screening process)
Evaluation of a Screening Programme
A programme is evaluated by asking:
- Does it reduce cause-specific mortality (not just survival)?
- What is the number needed to screen (NNS) to prevent one death?
- What is the gain in average life expectancy for the population?
Example: 731 women aged 65–69 must be screened by DEXA (and treated appropriately) to prevent one hip fracture from osteoporosis. — Harrison's, 22nd Ed.
Examples of Common Screening Programmes
| Disease | Screening Test | Target Group |
|---|---|---|
| Cervical cancer | Pap smear | Women 21–65 years |
| Breast cancer | Mammography | Women 40–74 years |
| Colorectal cancer | FOBT / Colonoscopy | Adults ≥45 years |
| Hypertension | Blood pressure measurement | All adults |
| Diabetes | Fasting blood glucose / HbA1c | Overweight, family history |
| Neonatal hypothyroidism | TSH (heel-prick) | All newborns |
| PKU | Guthrie test | All newborns |
| Tuberculosis | Mantoux / IGRA | High-risk populations |
| Glaucoma | Tonometry | Adults >40 years |
| Neural tube defects | Maternal serum AFP | Pregnant women |
Screening vs Diagnosis
| Feature | Screening | Diagnosis |
|---|---|---|
| Applied to | Apparently healthy population | Symptomatic individuals |
| Purpose | Detect presumptive cases | Confirm or exclude disease |
| Test type | Simple, cheap, rapid | Detailed, may be expensive |
| Result | Positive = needs further testing | Positive = definitive |
| Risk threshold | Lower (more FP acceptable) | Higher certainty required |
Key Formulas Summary
Sensitivity = TP / (TP + FN) × 100
Specificity = TN / (TN + FP) × 100
PPV = TP / (TP + FP) × 100
NPV = TN / (TN + FN) × 100
False Positive Rate = FP / (FP + TN) = 1 − Specificity
False Negative Rate = FN / (FN + TP) = 1 − Sensitivity
Sources: Harrison's Principles of Internal Medicine, 22nd Ed. | Textbook of Family Medicine, 9th Ed. | Emery's Elements of Medical Genetics and Genomics — supplemented with K. Park community medicine framework
Neonatal thermal protection op ghai short note
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
I have sufficient content from standard texts. OP Ghai's "Essential Pediatrics" is not in the library, but this is a well-defined neonatal topic. I'll now provide a comprehensive short note as per the OP Ghai framework.
Neonatal Thermal Protection
Short Note — As per OP Ghai's Essential Pediatrics
Introduction
Neonates are highly vulnerable to heat loss and hypothermia due to unique physiological characteristics. Thermal protection is a critical component of routine neonatal care and is a key part of the "warm chain" — the set of interlinked procedures performed at birth and in the newborn period to minimise heat loss.
Why Neonates Are Prone to Hypothermia
| Factor | Explanation |
|---|---|
| Large body surface area : weight ratio | Greater surface area for heat loss relative to body mass |
| Thin skin with little subcutaneous fat | Poor insulation, especially in preterm babies |
| Poor vasomotor control | Cannot constrict peripheral vessels efficiently |
| Cannot shiver effectively | Primary thermogenic response of adults is absent |
| Wet at birth | Rapid evaporative heat loss immediately after delivery |
| Head is proportionally large | Major source of radiant heat loss |
Normal Neonatal Temperature
| Parameter | Value |
|---|---|
| Normal axillary temperature | 36.5°C – 37.5°C |
| Mild hypothermia | 36.0°C – 36.4°C |
| Moderate hypothermia | 32.0°C – 35.9°C |
| Severe hypothermia | < 32.0°C |
Mechanisms of Heat Loss in Neonates
| Mechanism | Description | Example |
|---|---|---|
| Evaporation | Loss via water evaporation from wet skin | Wet baby at birth |
| Conduction | Direct contact with cold surfaces | Cold weighing scale, cold table |
| Convection | Heat loss to moving air currents | Draughts, open windows, fans |
| Radiation | Heat loss to cooler surrounding objects | Cold walls, cold incubator |
Evaporation is the most important immediately after birth.
The "Warm Chain" (10 Steps — WHO)
A series of interlinked procedures to prevent heat loss at and after birth:
- Warm delivery room — temperature ≥25°C (ideally 28°C)
- Warm resuscitation table — preheated with radiant warmer
- Immediate drying — dry the baby thoroughly with a warm cloth immediately after birth
- Skin-to-skin contact — place baby on mother's chest (kangaroo position), cover both with a blanket
- Breastfeeding — promotes warmth and metabolic stability
- Postpone bathing — do not bathe for at least 6 hours after birth (24 hours preferred); bathe only when temperature is stable
- Appropriate clothing and bedding — hat, socks, mittens, warm clothes; cover head (major heat loss area)
- Mother and baby together (rooming in) — skin-to-skin prevents hypothermia
- Warm transportation — keep warm during transport; use transport incubator or kangaroo care
- Training and awareness — train all birth attendants in thermal protection
Kangaroo Mother Care (KMC)
- Baby placed skin-to-skin on the mother's (or father's) chest between breasts
- Covered with a blanket or mother's clothing
- Especially important for low birth weight (LBW) and preterm babies
- Maintains temperature as effectively as an incubator
- Promotes breastfeeding, bonding, and reduces infection risk
- WHO strongly recommends KMC for all stable LBW infants
Prevention of Heat Loss at Birth — Immediate Steps
Following the "4 Warmths" concept at delivery:
- Warm the delivery room (≥25°C)
- Warm the receiving blanket/towel (pre-warmed)
- Dry immediately and thoroughly
- Cover head and body; place under radiant warmer
"Dry, Warm, Stimulate" — first three steps of neonatal resuscitation also prevent hypothermia. Wet blankets must be replaced immediately with dry, warm ones.
Management of Neonatal Hypothermia
Mild/Moderate Hypothermia:
- Skin-to-skin (KMC)
- Warm room, warm clothing
- Ensure breastfeeding or expressed breast milk feeds
- Radiant warmer / warm incubator
Severe Hypothermia:
- Radiant warmer (preferred in sick neonate)
- Warm IV fluids (if needed)
- Treat underlying cause (infection, hypoglycemia)
- Monitor temperature every 15–30 minutes until stable
- Rewarming rate: 0.5°C per hour (rapid rewarming causes apnoea and hypotension)
Consequences of Hypothermia in Neonates
- ↑ Oxygen consumption and metabolic rate → hypoxia
- Hypoglycaemia (glucose used up for thermogenesis)
- Metabolic acidosis
- Pulmonary vasoconstriction → respiratory distress
- Coagulation defects
- Intraventricular haemorrhage (in preterm)
- Increased mortality — especially in LBW infants
Neutral Thermal Environment (NTE)
The range of environmental temperature within which the neonate's metabolic rate is minimal and body temperature is maintained normally.
| Neonate | NTE |
|---|---|
| Term newborn (first days) | ~32–33°C |
| Preterm (<1500 g) | ~34–35°C |
| Incubator (general) | ~23°C ambient maintains core temp 36–37.5°C |
Key Points for Exam
- Most important immediate measure at birth: drying and wrapping
- Evaporation is the most important route of heat loss at birth
- KMC = most practical and effective thermal protection for LBW/preterm
- Do NOT bathe baby for at least 6 hours after birth
- Normal axillary temp: 36.5–37.5°C
- Neonates cannot shiver — they use non-shivering thermogenesis (brown adipose tissue/BAT)
- Brown fat (BAT) is located in: interscapular region, axillae, around kidneys and adrenals — metabolised to generate heat
Sources: Sabiston Textbook of Surgery | Rosen's Emergency Medicine | Harrison's Principles of Internal Medicine — compiled per OP Ghai Essential Pediatrics framework. Note: OP Ghai's textbook is not available in this library's collection.
Pathological jaundice,investigation and management of neonatal hyperbilirubinemia
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have sufficient content from authoritative sources. Let me compile a comprehensive answer on pathological neonatal jaundice.
Pathological Jaundice & Management of Neonatal Hyperbilirubinemia
(OP Ghai / Nelson Framework)
Bilirubin Metabolism in the Neonate
Bilirubin is produced from the breakdown of haemoglobin (80%) and other haem-containing proteins. In the neonate:
- Higher RBC mass + shorter RBC lifespan (70–90 days vs 120 days in adults) → more bilirubin produced
- Immature hepatic conjugation (low UGT1A1 enzyme activity)
- Increased enterohepatic circulation (high intestinal beta-glucuronidase activity deconjugates bilirubin back to unconjugated form)
Physiological vs Pathological Jaundice
| Feature | Physiological | Pathological |
|---|---|---|
| Onset | >24 hours after birth | <24 hours after birth |
| Duration | Resolves by day 7 (term), day 14 (preterm) | Persists >14 days (term) or >21 days (preterm) |
| Bilirubin rise | <5 mg/dL/day | >5 mg/dL/day |
| Peak bilirubin | <13 mg/dL (term), <15 mg/dL (preterm) | >13 mg/dL (term) at any time |
| Conjugated bilirubin | <2 mg/dL or <15% of total | >2 mg/dL (always pathological) |
| Clinical state | Baby looks well | May look unwell, pallor, hepatosplenomegaly |
Key rule: Jaundice appearing in the first 24 hours of life is ALWAYS pathological.
Causes of Pathological Jaundice
A. Unconjugated Hyperbilirubinemia
1. Haemolytic (most common cause of early jaundice <24 hrs):
- Isoimmune haemolysis — Rh incompatibility (anti-D), ABO incompatibility (most common)
- Hereditary spherocytosis, G6PD deficiency, pyruvate kinase deficiency
- Sepsis (acquired haemolysis)
2. Non-haemolytic:
- Polycythaemia (twin–twin transfusion, delayed cord clamping)
- Cephalhaematoma, bruising, swallowed blood
- Crigler-Najjar syndrome (Type I — complete UGT1A1 absence; Type II — partial)
- Gilbert syndrome
- Hypothyroidism
- Breast milk jaundice (late onset, peaks day 6–14, lasts weeks)
- Lucey-Driscoll syndrome (inhibitor of bilirubin conjugation in maternal serum)
B. Conjugated (Direct) Hyperbilirubinemia
Always pathological — indicates cholestasis or hepatocellular disease
- Biliary atresia (most important — surgical emergency)
- Neonatal hepatitis (TORCH infections — CMV, Rubella, Toxoplasma, Herpes)
- Choledochal cyst
- Alagille syndrome
- Metabolic — galactosaemia, alpha-1 antitrypsin deficiency, tyrosinaemia
- Total parenteral nutrition (TPN) cholestasis
- Sepsis (E. coli, staph)
Complications
Kernicterus (Bilirubin Encephalopathy)
Unconjugated bilirubin crosses the blood-brain barrier and deposits in:
- Basal ganglia (globus pallidus most affected)
- Subthalamic nuclei, hippocampus, brain stem nuclei
Risk factors for kernicterus:
- Prematurity (immature BBB)
- Hypoalbuminaemia (less bilirubin binding)
- Acidosis, hypoxia, sepsis (displace bilirubin from albumin)
- Haemolysis (rapid bilirubin rise)
Clinical stages of acute bilirubin encephalopathy:
| Stage | Features |
|---|---|
| Early | Lethargy, hypotonia, poor suck, high-pitched cry |
| Middle | Hypertonia, fever, opisthotonus, seizures |
| Late | Irreversible: deep stupor/coma |
Chronic kernicterus (Kernicteric tetrad):
- Choreoathetosis / dystonia
- Sensorineural hearing loss
- Upward gaze palsy (Parinaud's)
- Dental enamel dysplasia
Investigations
Mandatory workup for any neonate with jaundice:
| Investigation | Purpose |
|---|---|
| Total & direct serum bilirubin | Classify (unconjugated vs conjugated); severity |
| Blood group & Rh typing (mother & baby) | Detect isoimmune haemolysis |
| Direct Coombs test (DCT) | Positive in Rh/ABO incompatibility |
| Peripheral blood smear | Spherocytes (ABO incompatibility, hereditary spherocytosis), fragmented cells |
| Haemoglobin / haematocrit | Assess haemolysis/polycythaemia |
| Reticulocyte count | Elevated in haemolysis |
| Serum albumin | Determines free (unbound) bilirubin risk |
| Blood glucose | Hypoglycaemia worsens bilirubin toxicity |
| Sepsis screen (CBC, CRP, culture) | If infection suspected |
If conjugated hyperbilirubinemia:
- LFTs (ALT, AST, ALP, GGT)
- Urine reducing substances — galactosaemia
- Urine culture — UTI/sepsis
- TORCH serology
- Thyroid function (TSH, T4)
- Ultrasound abdomen — biliary atresia, choledochal cyst
- HIDA scan — biliary atresia (no excretion into gut)
- Liver biopsy — if biliary atresia suspected
Transcutaneous bilirubinometry (TcB):
- Non-invasive, useful for screening
- Needs confirmation with serum bilirubin if high or in high-risk neonates
Management
1. Phototherapy
Mechanism:
- Blue-green light (wavelength 430–490 nm, peak 460 nm) converts unconjugated bilirubin to:
- Lumirubin (most important product — excreted in bile/urine without conjugation)
- Configurational isomers (4Z,15E-bilirubin)
Indications (AAP guidelines):
- Based on age-specific nomograms (Bhutani curve) — bilirubin level vs postnatal age (hours)
- High-risk neonates (haemolysis, prematurity, sepsis) → lower threshold
- Generally: TSB ≥15 mg/dL at 24–48 hrs in term infant (varies by risk)
Technique:
- Special blue (narrow-band TL20) fluorescent lamps or LED phototherapy (most effective)
- Irradiance: ≥30 µW/cm²/nm for intensive phototherapy
- Expose maximum body surface area — cover eyes (retinal damage), shield gonads
- Continue feeds — increased fluid intake (↑ insensible losses)
- 4-hourly temperature monitoring
- Monitor bilirubin every 4–6 hours during intensive phototherapy
Side effects of phototherapy:
- Bronze baby syndrome (conjugated jaundice + phototherapy → grey-brown discolouration)
- Loose stools, skin rash
- Hyperthermia, dehydration
- Retinal damage (if eyes unshielded)
2. Exchange Transfusion (ET)
Indications:
- TSB ≥20–25 mg/dL in term neonates (age-specific thresholds)
- Rising bilirubin despite intensive phototherapy (>0.5 mg/dL/hr)
- Signs of acute bilirubin encephalopathy at any level
- Haemoglobin <10 g/dL with rapid rise in bilirubin (Rh disease)
Procedure:
- Double volume exchange = 2 × 85 mL/kg = ~160–170 mL/kg
- Removes ~85% of sensitised RBCs and ~50% of bilirubin
- Umbilical venous catheter (UVC) used
- Blood used: O-negative, CMV-negative, irradiated, leucodepleted packed RBCs mixed with FFP (to restore albumin)
- Alternating aliquots of 10 mL (preterm) or 20 mL (term) in/out
Complications of ET:
- Vascular — air embolism, thrombosis, NEC
- Haematological — thrombocytopenia, coagulopathy
- Metabolic — hypocalcaemia (citrate chelation), hypoglycaemia, acidosis
- Cardiovascular — arrhythmia, cardiac arrest
- Infectious — sepsis
- Graft vs host disease (if not irradiated blood)
- Mortality: ~0.3–0.5%
3. IV Immunoglobulin (IVIG)
- Indicated in Rh or ABO isoimmune haemolysis when TSB rising despite phototherapy
- Dose: 0.5–1 g/kg over 2 hours
- Mechanism: blocks Fc receptors on macrophages → reduces haemolysis
- Can reduce need for ET
4. Pharmacological
- Phenobarbitone — induces UGT1A1 (useful in Crigler-Najjar Type II, antenatal preparation)
- Tin-mesoporphyrin — inhibits haem oxygenase, reduces bilirubin production (investigational)
- Albumin infusion — 1 g/kg before ET to bind free bilirubin
5. Management of Conjugated Hyperbilirubinemia
- Biliary atresia — Kasai portoenterostomy (must be done before 60 days of age for best outcome)
- TORCH infections — specific antiviral therapy where available
- Galactosaemia — galactose-free diet
- Hypothyroidism — thyroxine replacement
- Sepsis — antibiotics
- Fat-soluble vitamin supplementation (A, D, E, K) in all cholestatic infants
Bhutani Nomogram (Risk Zones)
Bilirubin plotted against postnatal age in hours — divided into:
- High-risk zone (>95th percentile) — act immediately
- High-intermediate zone — close follow-up
- Low-intermediate zone
- Low-risk zone (<40th percentile)
Risk factors that lower treatment threshold: isoimmune haemolysis, G6PD deficiency, asphyxia, sepsis, acidosis, prematurity ≥35 weeks.
Summary Table: Management at a Glance
| Bilirubin Level (term, >72 hrs) | Action |
|---|---|
| <12 mg/dL | Observe, ensure feeds |
| 12–15 mg/dL | Investigate, consider phototherapy by risk |
| 15–20 mg/dL | Phototherapy |
| 20–25 mg/dL | Intensive phototherapy, prepare for ET |
| >25 mg/dL or encephalopathy signs | Exchange Transfusion |
Sources: Tietz Textbook of Laboratory Medicine, 7th Ed. | Creasy & Resnik's Maternal-Fetal Medicine | Rosen's Emergency Medicine — supplemented with OP Ghai Essential Pediatrics & AAP 2004/2022 guidelines framework.
Hemodynamics of atrial septal defect op ghai paediatrics short note
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Excellent — I now have comprehensive content and the diagram. Here is a complete short note.
Hemodynamics of Atrial Septal Defect (ASD)
Short Note — OP Ghai Essential Pediatrics
Definition
ASD is a congenital heart defect characterised by an abnormal, fixed opening in the atrial septum that allows communication between the left and right atria. It is one of the most common congenital heart defects, accounting for ~10% of all CHD, and is the most common CHD diagnosed in adults.
Types of ASD
| Type | Location | Frequency | Associations |
|---|---|---|---|
| Ostium Secundum | Central atrial septum (fossa ovalis) | ~70–75% | Usually isolated |
| Ostium Primum | Adjacent to AV valves (lower septum) | ~15–20% | Cleft mitral valve, VSD → part of AVSD |
| Sinus Venosus | Near SVC/IVC orifice | ~5–10% | Anomalous pulmonary venous drainage |
| Coronary sinus ASD | Coronary sinus unroofed | Rare | — |
ASD should be distinguished from Patent Foramen Ovale (PFO) — PFO is a normal fetal channel that fails to seal postnatally (present in ~20% of adults); it is not a fixed defect.
Embryological Basis
| Structure | Defect |
|---|---|
| Septum primum grows from posterior wall → leaves ostium primum anteriorly | Ostium primum ASD if septum primum fails to reach AV cushions |
| Septum primum develops fenestrations → forms ostium secundum | Ostium secundum ASD if too large or septum secundum too small |
| Septum secundum grows to cover ostium secundum, leaving foramen ovale | Closes at birth when LA pressure exceeds RA pressure |
Hemodynamics of ASD
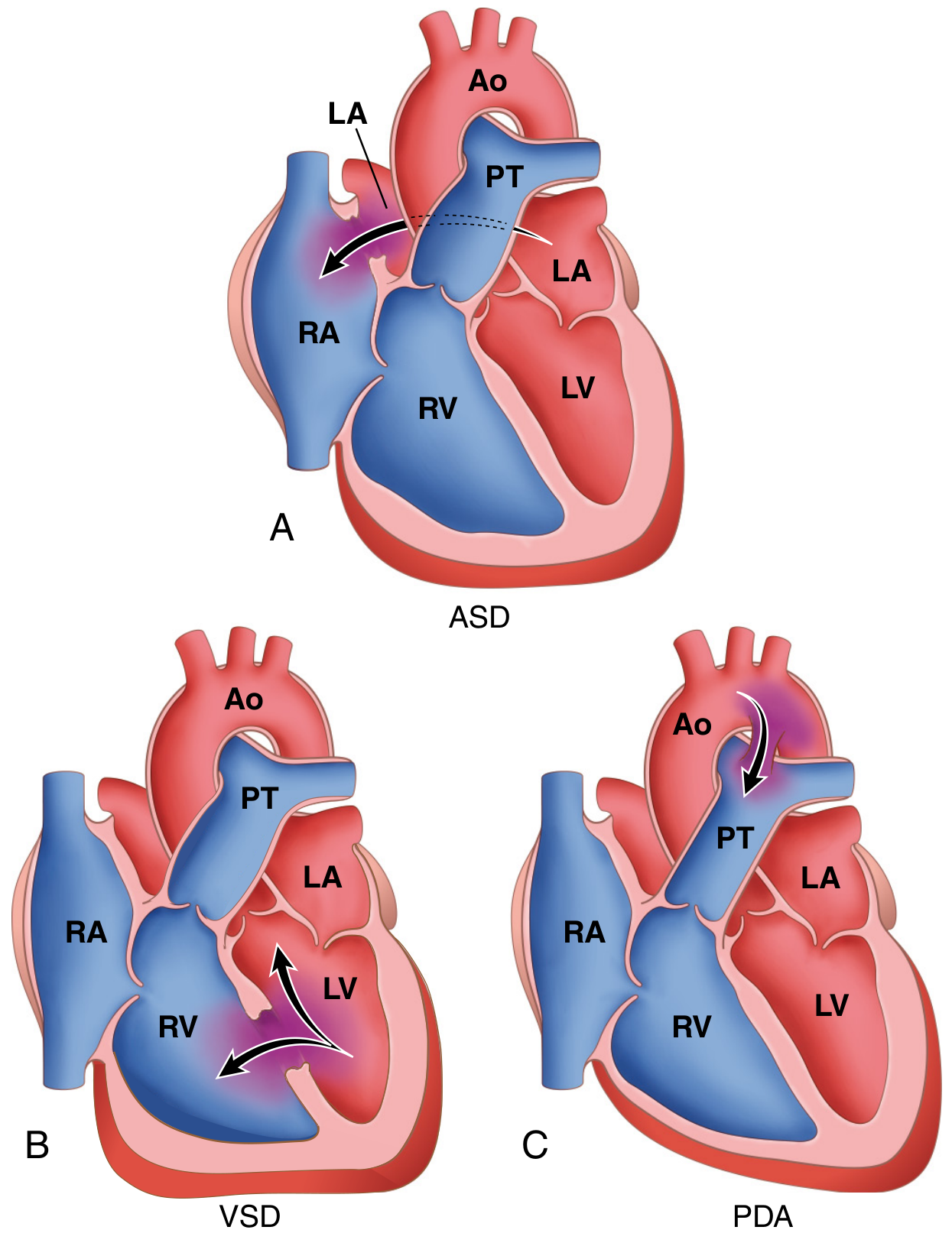
Why is the shunt Left → Right?
After birth:
- Pulmonary vascular resistance (PVR) falls → right-sided pressures decrease
- Left atrial pressure > Right atrial pressure (by ~5 mmHg)
- Right ventricle is more compliant (distensible) than the left → accepts extra volume more readily
- Net result: blood flows from LA → RA through the defect
Consequences of Left-to-Right Shunt:
LA → RA (through ASD)
↓
RV receives excess volume → RV volume overload / dilation
↓
Pulmonary artery flow increases (Qp:Qs = 2:1 to 8:1)
↓
Pulmonary vasculature handles increased flow
↓
Pulmonary plethora → progressive pulmonary hypertension (late)
Key Hemodynamic Points:
| Parameter | Change in ASD |
|---|---|
| Right atrium | Volume overloaded, dilated |
| Right ventricle | Volume overloaded, dilated (not hypertrophied initially) |
| Pulmonary artery | Dilated — increased flow |
| Left atrium | Normal or slightly reduced filling |
| Left ventricle | Normal size (blood diverted to RA before reaching LV) |
| Pulmonary blood flow (Qp) | Increased (2–8× normal) |
| Systemic blood flow (Qs) | Normal or slightly reduced |
| Qp:Qs ratio | >1.5:1 = significant shunt; >2:1 = surgery indicated |
Why is there no early pulmonary hypertension?
- The shunt is at low pressure (atrial level) — pressure difference between LA and RA is small (~5 mmHg)
- The pulmonary vasculature can accommodate increased volume flow for many years
- Pulmonary hypertension and Eisenmenger syndrome occur late (usually adulthood, 3rd–5th decade)
Clinical Features
Symptoms:
- Usually asymptomatic in childhood — the hallmark of ASD
- Fatigue, exertional dyspnoea (if large shunt)
- Recurrent respiratory tract infections (increased pulmonary blood flow)
- Rarely: cardiac failure in infancy (large defects)
- Adults: palpitations, AF, stroke (paradoxical embolism via PFO)
Signs:
| Finding | Explanation |
|---|---|
| Wide, fixed splitting of S2 | Most characteristic sign — RV stroke volume unchanged during respiration because RA always has extra blood from ASD; A2 early + P2 delayed and fixed |
| Pulmonary ejection systolic murmur (grade 2–3/6, left 2nd ICS) | Due to increased flow across pulmonary valve (not turbulence through ASD itself) |
| Tricuspid mid-diastolic rumble (lower left sternal border) | Increased flow across tricuspid valve |
| Right ventricular heave | RV volume overload |
| Widely split S2 does NOT vary with breathing | Key differentiator from normal splitting |
The murmur of ASD is NOT from the ASD itself — it is a flow murmur across the pulmonary valve due to increased Qp.
Investigations
ECG:
| Type | ECG Pattern |
|---|---|
| Ostium Secundum ASD | Right axis deviation, rSR' pattern (incomplete RBBB) in V1, right atrial enlargement |
| Ostium Primum ASD | Left axis deviation (superior QRS axis), RBBB, prolonged PR |
Chest X-Ray:
- Cardiomegaly (RV and RA enlargement)
- Pulmonary plethora (prominent pulmonary vasculature)
- Dilated pulmonary artery trunk
- Small aortic knuckle
Echocardiography (Investigation of Choice):
- 2D Echo: visualise defect size and location
- Colour Doppler: shows left-to-right shunt across atrial septum
- Bubble contrast echo: confirms shunt
- Estimate Qp:Qs ratio
- Assess RV size and function
Cardiac catheterisation (rarely needed):
- Step-up in oxygen saturation at RA level (oxygenated LA blood mixing)
- Measures Qp:Qs precisely
- Measures PVR before surgery in suspected pulmonary hypertension
Complications
- Pulmonary arterial hypertension (late, after decades)
- Eisenmenger syndrome — shunt reversal (R→L), cyanosis, polycythaemia (rare in ASD, occurs late in adulthood)
- Atrial arrhythmias — AF, atrial flutter (due to RA dilation)
- Paradoxical embolism → stroke/TIA
- Right heart failure
- Infective endocarditis (uncommon in isolated secundum ASD)
Management
Indications for Closure:
- Qp:Qs ≥1.5:1 with symptoms or evidence of RV volume overload
- Any symptomatic ASD
- Evidence of paradoxical embolism
- Optimal age: 3–5 years (before school age, before pulmonary hypertension)
- Contraindicated if PVR >8 Wood units (Eisenmenger physiology)
Methods:
| Method | Indication |
|---|---|
| Transcatheter device closure (Amplatzer occluder) | Ostium secundum ASD <38 mm with adequate rim — method of choice |
| Surgical repair (cardiopulmonary bypass) | Primum ASD, sinus venosus ASD, large secundum not suitable for device |
Spontaneous closure:
- Secundum ASD <8 mm may close spontaneously in the first 2 years of life
- Large defects and primum/sinus venosus ASDs do not close spontaneously
Summary: Hemodynamic Sequence
Atrial septal defect
↓
LA pressure > RA pressure
↓
Left-to-right shunt (LA → RA)
↓
↑ RA volume → ↑ RV volume → ↑ Pulmonary flow (Qp ↑)
↓
RV dilation + PA dilation + pulmonary plethora
↓
Wide FIXED splitting of S2 + Pulmonary flow murmur
↓
Late: Pulmonary hypertension → Eisenmenger (reversal)
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease, 10th Ed. | Creasy & Resnik's Maternal-Fetal Medicine — compiled per OP Ghai Essential Pediatrics framework.
Urinary tract infection in children-clinical features ,investigation,management,management of recurrent OTI
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
I now have authoritative content from Red Book 2021 on UTI chemoprophylaxis in children. Combined with standard OP Ghai/Nelson content I know comprehensively, here is a detailed answer.
Urinary Tract Infection (UTI) in Children
Clinical Features, Investigation, Management & Recurrent UTI
(OP Ghai / Nelson Pediatrics Framework)
Definition & Classification
UTI = significant bacteriuria (>10⁵ CFU/mL of a single organism on midstream clean-catch urine) with clinical symptoms AND/OR pyuria.
| Type | Definition |
|---|---|
| Cystitis | Lower UTI — infection confined to bladder/urethra |
| Pyelonephritis | Upper UTI — infection involving renal parenchyma; fever, loin pain |
| Asymptomatic bacteriuria | Significant bacteriuria without symptoms (no treatment in children usually) |
| First UTI | First documented episode |
| Recurrent UTI | ≥2 episodes of febrile UTI, or ≥3 episodes of lower UTI |
Epidemiology
- Overall prevalence: ~3–5% in girls, ~1% in boys
- In the neonatal/infant period: more common in boys (due to anatomical phimosis)
- After infancy: girls >> boys (short urethra, perineal colonisation)
- ~30–40% of children with first UTI have vesicoureteral reflux (VUR)
- Uncircumcised boys have 10× higher risk than circumcised
Aetiology / Causative Organisms
| Organism | Frequency |
|---|---|
| Escherichia coli | 75–90% (most common) |
| Klebsiella pneumoniae | 5–10% |
| Proteus mirabilis | Common in boys (urease producer → struvite stones) |
| Staphylococcus saprophyticus | Adolescent girls |
| Enterococcus faecalis | Neonates, hospitalised |
| Pseudomonas aeruginosa | Catheterised / instrumented patients |
Pathogenesis & Predisposing Factors
Ascending route (most common):
Perineal/periurethral colonisation → bladder → ureter → kidney
Predisposing factors:
| Category | Factors |
|---|---|
| Structural | VUR, pelviureteric junction (PUJ) obstruction, posterior urethral valves (PUV), duplex system, ureterocele, horseshoe kidney |
| Functional | Bladder-bowel dysfunction (BBD), neurogenic bladder, voiding dysfunction |
| Host | Female sex, uncircumcised male, constipation, pinworm infestation, sexual abuse |
| Neonatal | Haematogenous spread; prematurity |
Clinical Features
Age-dependent presentation:
| Age Group | Symptoms |
|---|---|
| Neonates (<1 month) | Non-specific: fever/hypothermia, poor feeding, vomiting, jaundice (prolonged conjugated), lethargy, sepsis picture |
| Infants (1–24 months) | Fever without focus (most common presentation), irritability, vomiting, poor weight gain, foul-smelling urine |
| Older children (>2 years) | Dysuria, frequency, urgency, suprapubic pain (cystitis); fever, loin/flank pain, rigors, vomiting (pyelonephritis) |
| Adolescents | Cystitis — dysuria, frequency, urgency; pyelonephritis — high fever, costovertebral angle tenderness |
Signs:
- Fever (>38°C) — especially in upper UTI
- Suprapubic tenderness (cystitis)
- Costovertebral angle (CVA) tenderness / loin pain (pyelonephritis)
- Abdominal distension (especially neonates)
- In severe cases: signs of urosepsis (shock, tachycardia)
Investigations
1. Urine Analysis & Culture (cornerstone)
Urine collection methods (reliability hierarchy):
| Method | Age | Contamination risk |
|---|---|---|
| Suprapubic aspiration (SPA) | <2 years | Lowest — gold standard in infants |
| Urethral catheterisation | Infants/young children | Low |
| Midstream clean catch (MSCC) | Older toilet-trained children | Moderate |
| Bag urine specimen | Only screening; NOT for culture | High (50–80% contamination) |
Diagnostic criteria:
| Parameter | Significant |
|---|---|
| Colony count | >10⁵ CFU/mL (MSCC), >10⁴ (catheter), any growth (SPA) |
| Pyuria | >5 WBC/hpf on microscopy; positive leukocyte esterase on dipstick |
| Nitrites | Positive (suggests gram-negative organisms) |
| Bacteriuria | Presence of bacteria on Gram stain |
Sensitivity: Pyuria + nitrites combined = ~85–90% for UTI Urine culture is mandatory before starting antibiotics in all children
2. Blood Investigations:
- CBC — leucocytosis (pyelonephritis / sepsis)
- CRP / Procalcitonin — elevated in upper UTI; helps distinguish pyelonephritis from cystitis
- Blood culture — mandatory in neonates, infants <3 months, and ill-looking children (bacteraemia in 4% of febrile UTI)
- Serum creatinine / BUN — assess renal function
- Serum electrolytes
3. Imaging (crucial for structural evaluation):
| Investigation | When | Purpose |
|---|---|---|
| Renal USG | All children with first UTI, done promptly | Detect hydronephrosis, structural anomalies, abscess, renal size |
| MCUG (Micturating cystourethrogram) | After first febrile UTI in <2 yrs, or recurrent UTI, or USG abnormality | Diagnose VUR (gold standard), detect PUV, bladder abnormalities |
| DMSA scan (Tc-99m dimercaptosuccinic acid) | Acute phase: cortical defects (acute pyelonephritis); 4–6 months later: permanent scarring | Most sensitive for renal scarring; localises upper UTI |
| MAG3/DTPA renogram | Obstruction suspected | Assess drainage + differential renal function |
| Intravenous urogram (IVU) | Less used now; replaced by USG+DMSA | — |
AAP/NICE Imaging algorithm (simplified):
- Well-responding febrile UTI in 2–24 months: renal USG if not responding in 48 hrs or at completion of treatment
- Abnormal USG or recurrent UTI: MCUG + DMSA
Management
General Principles:
- Start antibiotic after urine sample is collected (do not delay for culture results in sick children)
- Adequate fluid intake
- Treat constipation and voiding dysfunction if present
A. Acute Management
Cystitis (Lower UTI — afebrile):
- Oral antibiotics for 3–7 days
- First-line agents:
- Nitrofurantoin (not <3 months, avoid if renal impairment)
- Trimethoprim (TMP) or co-trimoxazole (TMP-SMX)
- Cephalexin / Cefixime
- Amoxicillin-clavulanate (if organism sensitive)
- Avoid amoxicillin alone (high E. coli resistance)
Pyelonephritis (Upper UTI — febrile):
Outpatient oral (if >3 months, not toxic, able to tolerate orals):
- Oral cefixime or co-amoxiclav for 10–14 days
- Ceftibuten, ciprofloxacin (older children)
Inpatient IV (neonates, infants <3 months, toxic-looking, vomiting, failed oral, urosepsis):
- IV Ampicillin + Gentamicin (neonates)
- IV Ceftriaxone / Cefotaxime (infants and children)
- Switch to oral after 48–72 hrs of clinical improvement
Duration: 10–14 days total for febrile UTI / pyelonephritis
B. Follow-Up After Treatment
- Urine culture 48–72 hrs after starting antibiotics (test of cure if not improving)
- Repeat culture 1 week after completion to confirm eradication
- Imaging as per protocol
Vesicoureteral Reflux (VUR) — Key Complication
| Grade | Description |
|---|---|
| I | Reflux into ureter only |
| II | Reflux into ureter + pelvis, no dilation |
| III | Mild dilation of ureter and pelvis |
| IV | Moderate dilation, blunting of fornices |
| V | Gross dilation, tortuosity, loss of papillary impressions |
Reflux nephropathy = renal scarring from recurrent infected VUR → chronic kidney disease, hypertension.
Management of Recurrent UTI
Recurrent UTI = ≥2 febrile UTIs, or ≥3 afebrile UTIs.
Step 1: Identify and correct underlying cause
- Investigate with USG, MCUG, DMSA
- Treat constipation (very common — impairs bladder emptying)
- Bladder-bowel dysfunction (BBD): timed voiding, double voiding, laxatives
- Correct structural anomalies surgically if indicated (PUJ obstruction, PUV)
Step 2: Antibiotic Prophylaxis (Continuous Low-Dose Prophylaxis — CAP)
Indications (controversial — use selectively):
- Children with high-grade VUR (Grade III–V) — strongest evidence
- Children with structural abnormalities causing urinary stasis
- Children <1 year with any grade VUR (pending resolution)
- Not recommended routinely for children with low-grade VUR or no VUR (NICE/AAP 2011)
RIVUR Study (2014): TMP-SMX prophylaxis reduced recurrent UTI by 50% in children with Grade I–IV VUR, but did not reduce renal scarring and increased antibiotic resistance (from 25% → 68%). — Red Book 2021
Prophylaxis agents (given at 25–30% of therapeutic dose, once nightly):
| Drug | Dose | Notes |
|---|---|---|
| Trimethoprim | 1–2 mg/kg once at night | First-line; avoid <6 weeks |
| Co-trimoxazole (TMP-SMX) | TMP 2 mg/kg at night | First-line in many centres |
| Nitrofurantoin | 1–2 mg/kg at night | Avoid if GFR low; avoid <3 months |
| Cephalexin | 10–15 mg/kg at night | Used in neonates/infants <3 months |
| Nalidixic acid | Not commonly used now | — |
Duration of prophylaxis:
- Until VUR resolves (confirmed on follow-up MCUG at 1–2 years)
- Until age 5 years in anatomical anomalies
- Re-evaluate yearly
Step 3: Surgical Management (for VUR)
| Approach | Indications |
|---|---|
| Endoscopic injection (STING procedure) — subureteric Deflux injection | Grade II–IV VUR; minimally invasive |
| Open/laparoscopic ureteral reimplantation (Cohen/Politano-Leadbetter) | Grade IV–V, failed endoscopic, anatomical abnormalities |
Step 4: General Measures for Recurrent UTI
- Adequate hydration — dilutes bacteria, promotes bladder washout
- Regular and timed voiding (every 2–3 hours) — avoid infrequent voiding
- Double voiding — void, wait 5 minutes, void again (empties bladder fully)
- Treat constipation aggressively (lactulose, bowel training)
- Perineal hygiene — front-to-back wiping in girls
- Avoid bubble baths, tight clothing
- Circumcision in boys with recurrent UTI and high-grade VUR (reduces recurrence)
- Cranberry juice — some evidence in older girls (not routinely recommended in young children)
Complications of Untreated/Recurrent UTI
- Renal scarring (reflux nephropathy) → commonest serious complication
- Hypertension (renovascular)
- Chronic kidney disease / end-stage renal disease
- Urosepsis / septicaemia (especially in neonates)
- Renal abscess
Summary Table: Management at a Glance
| Situation | Action |
|---|---|
| Neonates / Infants <3 months | IV antibiotics (Ampicillin + Gentamicin); admission mandatory |
| Febrile UTI (>3 months), not toxic | Oral cefixime/co-amoxiclav for 10–14 days |
| Cystitis (afebrile, older child) | Oral TMP or nitrofurantoin for 3–7 days |
| VUR Gr III–V or structural anomaly | CAP (TMP or nitrofurantoin) |
| High-grade VUR failing prophylaxis | STING injection or ureteric reimplantation |
| Constipation / voiding dysfunction | Treat aggressively — reduces recurrence significantly |
Sources: Red Book 2021 — Report of the Committee on Infectious Diseases (AAP) | Campbell-Walsh Urology | Comprehensive Clinical Nephrology, 7th Ed. — supplemented with OP Ghai Essential Pediatrics / Nelson Textbook framework.
Status epilipticus criteria and treatment
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Excellent — I have comprehensive content from multiple authoritative sources and the treatment algorithm. Here is a complete, exam-ready answer.
Status Epilepticus (SE) — Criteria & Treatment
Definition & Diagnostic Criteria
Traditional definition (historical):
Status epilepticus = seizure lasting >30 minutes, OR two or more sequential seizures without full recovery of consciousness between them.
Current operational definition (ILAE 2015 / clinical practice):
| Seizure Type | Treat when duration reaches | Likely neuronal injury if untreated > |
|---|---|---|
| Generalised tonic-clonic SE | ≥5 minutes | 30 minutes |
| Focal impaired awareness SE | ≥10 minutes | 60 minutes |
| Absence SE | ≥10–15 minutes | — |
The shift from 30 minutes to 5 minutes as the operational treatment threshold reflects the fact that most seizures self-terminate within 2–3 minutes; any seizure persisting beyond 5 minutes is unlikely to stop spontaneously and requires treatment. — Katzung's Pharmacology, 16th Ed.; Goldman-Cecil Medicine
Classification
By Convulsive Activity:
| Type | Features |
|---|---|
| Convulsive SE (CSE) | Bilateral motor activity (tonic-clonic) + impaired consciousness — most common, most dangerous |
| Non-convulsive SE (NCSE) | Persistent altered consciousness/behaviour without major motor signs; confirmed on EEG; includes absence SE, focal SE, NCSE in coma |
| Subtle SE | Motor activity ceases (after prolonged CSE) but EEG continues — electromechanical dissociation; if patient doesn't wake within 30 min of stopping motor activity, suspect this |
By Treatment Response:
| Phase | Definition |
|---|---|
| Early SE | 5–10 minutes |
| Established SE | 10–30 minutes |
| Refractory SE (RSE) | Seizures continue/recur ≥30 minutes after first- and second-line treatment |
| Super-refractory SE (SRSE) | Continues ≥24 hours after anaesthetic agents, including recurrence on tapering |
Aetiology / Causes
| Category | Examples |
|---|---|
| Acute symptomatic | Stroke, TBI, CNS infection (meningitis, encephalitis — most common cause overall), hypoxic-ischaemic encephalopathy, metabolic (hypoglycaemia, hyponatraemia, hypocalcaemia), drug toxicity, alcohol withdrawal |
| Remote symptomatic | Prior stroke, TBI, CNS surgery |
| Progressive | Brain tumour, autoimmune encephalitis (NMDAR, LGI1) |
| Unknown | ~50% remain cryptogenic even after full evaluation |
| Febrile SE | In children: prolonged febrile seizures |
| Epilepsy-related | Sub-therapeutic AED levels, non-compliance |
Treatment Algorithm
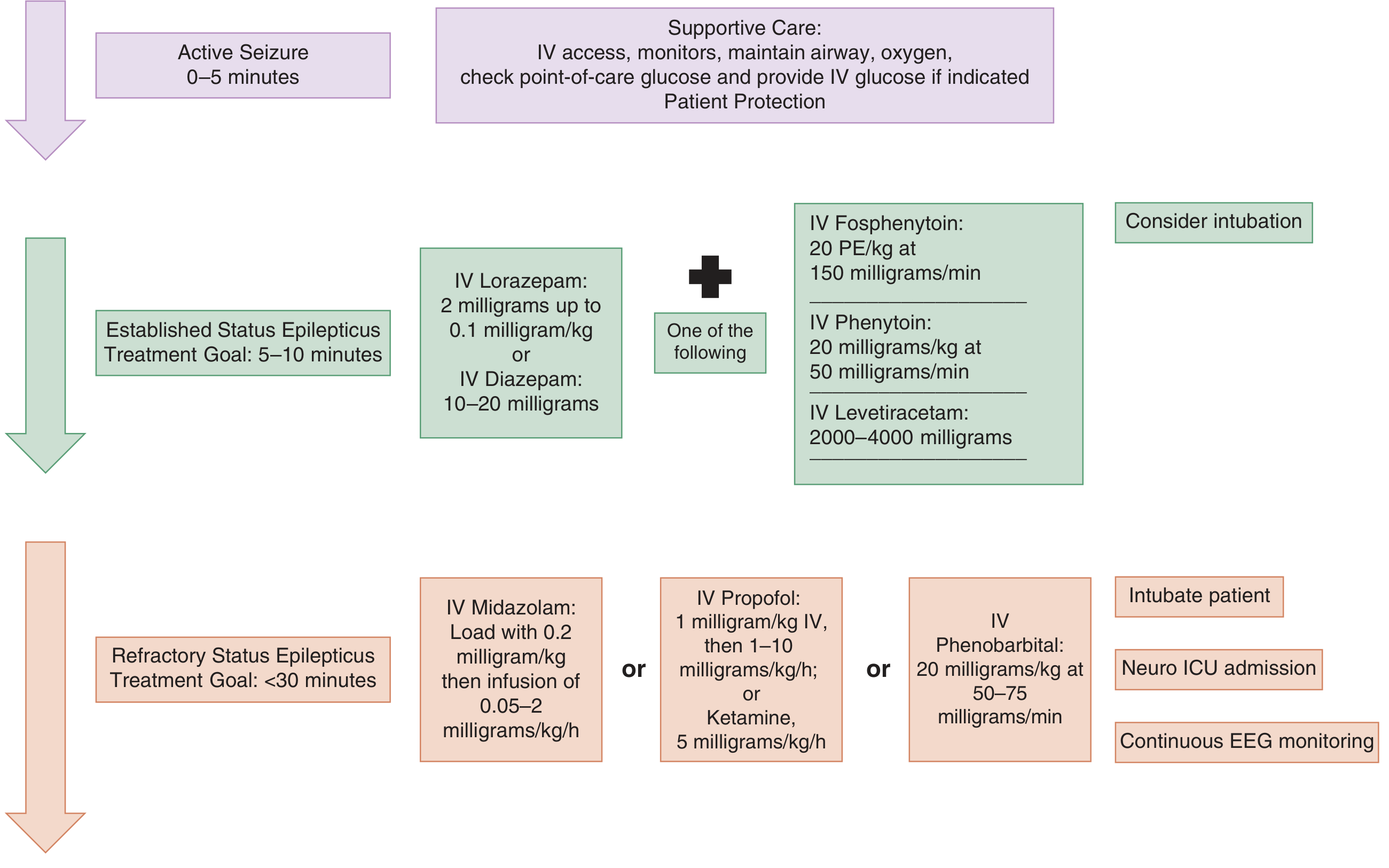
PHASE 0 (0–5 min): Active Seizure — Stabilisation
Simultaneous with treatment initiation:
- Airway — position patient, airway adjunct if needed
- Breathing — oxygen (high-flow), pulse oximetry, monitor SpO₂
- Circulation — large-bore IV access × 2, cardiac monitor, BP monitoring
- Glucose — bedside blood glucose immediately; if <60 mg/dL or unknown → 50 mL 50% dextrose IV (preceded by thiamine 100 mg IV if alcoholism/malnutrition suspected)
- Position patient in left lateral position — prevent aspiration
- Labs: CBC, CMP (Na, Ca, Mg, glucose), creatinine, LFTs, AED levels, toxicology screen, ABG, blood cultures if infection suspected
- ECG monitoring
PHASE 1 (5–10 min): FIRST-LINE — Benzodiazepines
Goal: Stop seizure within 5–10 minutes of treatment
| Route | Drug | Dose | Onset | Duration |
|---|---|---|---|---|
| IV | Lorazepam (preferred) | 0.1 mg/kg IV (adult: 2–4 mg); may repeat once | ~2–3 min | 12–24 hrs |
| IV | Diazepam | 5–10 mg IV at 5 mg/min | ~2 min | 15–60 min |
| IM | Midazolam (if no IV access) | 10 mg IM (adult >40 kg); 5 mg if 13–40 kg | ~5 min | 1–2 hrs |
| Rectal | Diazepam gel | 0.2–0.5 mg/kg (children) | ~5 min | Short |
| Intranasal / Buccal | Midazolam | 0.2–0.5 mg/kg (children) | ~5 min | Short |
Key pharmacology:
- Lorazepam: less lipophilic → slower redistribution from brain → longer effective duration than diazepam
- IM midazolam = as effective as IV lorazepam in prehospital setting (RAMPART trial)
- Can repeat benzodiazepine once if no response in 5 minutes
- Risk: respiratory depression — have bag-mask ventilation ready
Efficacy: ~70% of SE controlled with first-line benzodiazepines alone
PHASE 2 (10–30 min): SECOND-LINE — Established SE
Start within 20 minutes of diagnosis. All four agents are equally effective (ESETT trial 2019 — no significant difference in outcomes).
| Drug | Dose | Rate | Notes |
|---|---|---|---|
| Fosphenytoin (preferred over phenytoin) | 20 PE/kg IV | 150 mg PE/min | Water-soluble prodrug; fewer infusion-site reactions; can give IM; monitor BP/cardiac |
| Phenytoin | 20 mg/kg IV | Max 50 mg/min (25 mg/min safer) | Incompatible with glucose solutions; cardiac monitoring mandatory; hypotension, arrhythmia risk |
| Levetiracetam | 30–60 mg/kg IV (adult: 2000–4500 mg) | Over 10–15 min | Safest cardiac profile; no hepatic interactions; preferred in liver disease, pregnancy |
| Valproate | 30–40 mg/kg IV | 5 mg/kg/min | Avoid in liver disease, mitochondrial disorders, pregnancy (teratogenic); effective |
| Lacosamide | 200–400 mg IV | 15–60 min | Second-line alternative; sodium channel slow inactivation |
| Phenobarbitone | 15–20 mg/kg IV | 100 mg/min | Effective but sedating; used in neonates as first-line |
Consider intubation at this phase if airway protection is needed or if patient is deteriorating.
PHASE 3 (>30 min): REFRACTORY SE — ICU Management
All patients require:
- Endotracheal intubation and mechanical ventilation
- Continuous EEG monitoring (to detect NCSE, guide drug titration)
- Neuro-ICU admission
Anaesthetic/continuous infusion agents — titrate to burst suppression on EEG:
| Drug | Loading Dose | Infusion | Notes |
|---|---|---|---|
| Midazolam (first-line) | 0.2 mg/kg IV bolus | 0.05–2 mg/kg/hr | Tachyphylaxis common; titrate up |
| Propofol | 1–2 mg/kg IV | 1–15 mg/kg/hr | Propofol infusion syndrome risk (>48 hrs, high dose); monitor triglycerides, pH |
| Pentobarbital | 5–15 mg/kg IV | 0.5–5 mg/kg/hr | Deepest sedation; hypotension common; requires vasopressors |
| Thiopental | 3–5 mg/kg IV | Continuous | Similar to pentobarbital |
| Ketamine | 1–2 mg/kg IV bolus | 2.2–5 mg/kg/hr | NMDA antagonist; useful in super-refractory SE; minimal cardiovascular depression |
Therapeutic hypothermia is NOT beneficial in SE. — Goldman-Cecil Medicine
Super-Refractory SE (>24 hrs on anaesthetics):
Additional options:
- Ketogenic diet (via NG tube)
- Pyridoxine (in neonates/infants — B6-dependent SE)
- Immunotherapy — IV steroids, IVIG, plasma exchange (if autoimmune encephalitis)
- Electroconvulsive therapy (ECT) — case reports
- Vagal nerve stimulation
- Surgical resection (if focal structural lesion)
- Transcranial magnetic stimulation (TMS)
Summary of Drug Doses (Adults)
| Phase | Drug | Dose |
|---|---|---|
| 1st line | IV Lorazepam | 0.1 mg/kg (up to 4 mg); repeat once |
| 1st line (no IV) | IM Midazolam | 10 mg IM |
| 2nd line | IV Fosphenytoin | 20 PE/kg at 150 mg PE/min |
| 2nd line | IV Levetiracetam | 2000–4500 mg over 10–15 min |
| 2nd line | IV Valproate | 30–40 mg/kg at 5 mg/kg/min |
| 3rd line (RSE) | IV Midazolam infusion | 0.2 mg/kg load → 0.05–2 mg/kg/hr |
| 3rd line (RSE) | IV Propofol | 1–2 mg/kg load → 1–15 mg/kg/hr |
| 3rd line (RSE) | IV Pentobarbital | 5–15 mg/kg load → 0.5–5 mg/kg/hr |
In Children (Paediatric Specifics — OP Ghai)
| Phase | Drug | Dose |
|---|---|---|
| 1st line | Rectal/IV Diazepam | 0.5 mg/kg rectal; 0.3 mg/kg IV |
| 1st line (preferred) | Buccal/IM Midazolam | 0.2–0.5 mg/kg buccal; 0.2 mg/kg IM |
| 1st line IV | Lorazepam | 0.1 mg/kg IV |
| 2nd line | IV Phenobarbitone | 20 mg/kg IV at 1 mg/kg/min |
| 2nd line | IV Phenytoin/Fosphenytoin | 20 mg/kg IV at 1 mg/kg/min |
| 2nd line | IV Levetiracetam | 30–60 mg/kg IV |
| 3rd line | IV Midazolam infusion | 0.1–0.4 mg/kg/hr |
| Neonatal SE | IV Phenobarbitone (1st line) | 20 mg/kg; then phenytoin |
Monitoring & Investigations During Management
- Continuous EEG — essential once paralysis given; confirms NCSE; guides anaesthetic titration
- CT head — after seizure control (if new-onset SE, focal signs, trauma)
- LP — after seizure control if CNS infection suspected (empiric antibiotics/antivirals should NOT be delayed)
- MRI brain — once stabilised
- Temperature monitoring — treat hyperthermia actively (worsens neuronal injury)
Prognosis
- Mortality: ~10–20% overall; up to 40% in RSE
- Neurological sequelae in survivors of prolonged SE: cognitive impairment, new-onset epilepsy
- Outcome determined primarily by underlying aetiology, not duration alone
Sources: Goldman-Cecil Medicine, International Edition | Tintinalli's Emergency Medicine, 9th Ed. | Katzung's Basic & Clinical Pharmacology, 16th Ed. | Bradley and Daroff's Neurology in Clinical Practice
Hyperkalemia investigation and management
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
I now have comprehensive, authoritative data from Harrison's, Tintinalli's, Miller's Anesthesia, and Rosen's EM. Here is a complete answer.
Hyperkalemia — Investigation & Management
Definition & Classification
Hyperkalemia = serum potassium >5.5 mEq/L
| Grade | Serum K⁺ | Clinical Significance |
|---|---|---|
| Mild | 5.5–6.0 mEq/L | Often asymptomatic |
| Moderate | 6.0–6.5 mEq/L | ECG changes may appear |
| Severe | >6.5 mEq/L | Life-threatening; immediate treatment required |
| Critical | >7.0–8.0 mEq/L | Cardiac arrest risk |
Always exclude pseudohyperkalemia first (haemolysis during phlebotomy — most common in infants/children; prolonged tourniquet, thrombocytosis, leucocytosis). Repeat sample from a free-flowing vein.
Normal Potassium Physiology
- Total body K⁺ ~3500 mEq; 98% is intracellular (mainly muscle)
- Serum K⁺ is maintained at 3.5–5.0 mEq/L
- Kidney excretes 90–95% of daily K⁺ load via aldosterone-stimulated principal cells of the cortical collecting duct (Na⁺/K⁺ exchange)
- Insulin, catecholamines (β₂), and alkalosis drive K⁺ intracellularly
- Acidosis, cell lysis, and hyperosmolarity shift K⁺ extracellularly
Causes of Hyperkalemia
1. Excess Intake / Exogenous Load
- Excessive K⁺ supplementation (IV or oral)
- Massive blood transfusion (stored blood K⁺ leaks from RBCs)
- Salt substitutes (KCl-containing)
2. Transcellular Shift (K⁺ out of cells)
- Metabolic acidosis — H⁺ enters cells, K⁺ exits (each 0.1 fall in pH → ~0.6 mEq/L rise in K⁺)
- Insulin deficiency / DKA — insulin normally drives K⁺ into cells
- β-blockers — block β₂-mediated cellular K⁺ uptake
- Hyperosmolarity — draws water and K⁺ out of cells
- Cell destruction / lysis:
- Rhabdomyolysis
- Tumour lysis syndrome
- Haemolytic anaemia
- Major trauma, burns
- Massive haematoma
3. Decreased Renal Excretion (most common cause)
- Acute kidney injury (AKI) — especially oliguric
- Chronic kidney disease (CKD) — most common cause overall
- Hypoaldosteronism:
- Addison's disease (primary adrenal insufficiency)
- Hyporeninemic hypoaldosteronism (common in diabetes, elderly, CKD)
- Congenital adrenal hyperplasia (salt-wasting type)
- Distal tubular dysfunction: sickle cell, amyloidosis, obstructive uropathy
4. Drugs (very common)
| Drug | Mechanism |
|---|---|
| ACE inhibitors / ARBs | ↓ Angiotensin II → ↓ Aldosterone |
| Potassium-sparing diuretics (spironolactone, amiloride, triamterene) | Block aldosterone or ENaC |
| NSAIDs | ↓ Renin → ↓ Aldosterone |
| Heparin | Inhibits aldosterone synthesis |
| Trimethoprim / Pentamidine | Block ENaC (like amiloride) |
| Digoxin toxicity | Blocks Na⁺/K⁺-ATPase |
| Succinylcholine | Depolarising agent → K⁺ efflux |
| Beta-blockers | Block β₂-mediated K⁺ uptake |
Clinical Features
Neuromuscular:
- Muscle weakness (ascending) → flaccid paralysis (in severe cases)
- Paraesthesias
- Fatigue, malaise
- Rarely: respiratory muscle paralysis
Cardiac (most dangerous — from altered membrane potential):
- Palpitations, chest discomfort
- Bradycardia, arrhythmias
- Cardiac arrest (VF or asystole)
GI:
- Nausea, vomiting, abdominal cramping, diarrhoea
ECG Changes in Hyperkalemia
ECG changes correlate with K⁺ level and are the key guide to urgency of treatment:
| Serum K⁺ | ECG Change |
|---|---|
| 5.5–6.5 mEq/L | Tall, peaked (tented) T waves — narrow base, symmetric; shortened QT interval |
| 6.5–7.5 mEq/L | Prolonged PR interval; flattening/disappearance of P waves |
| 7.0–8.0 mEq/L | Widened QRS complex (bundle branch block pattern) |
| >9.0 mEq/L | "Sine wave" pattern (merging of P, QRS, T) → VF → asystole |
Any ECG change = cardiac emergency → treat immediately with calcium
The ECG is more important than the absolute K⁺ level in determining urgency.
Investigation
Confirm & Grade:
- Repeat serum potassium (to exclude pseudohyperkalemia) — free-flowing venipuncture without tourniquet
- ECG — mandatory; guides urgency
Identify Cause:
| Test | Purpose |
|---|---|
| Serum creatinine / BUN / eGFR | Renal failure (most common cause) |
| Serum glucose | DKA / insulin deficiency |
| Arterial blood gas / serum bicarbonate | Metabolic acidosis |
| Serum Na, Cl (anion gap) | Metabolic context |
| Serum Ca, Mg, PO₄ | Related electrolytes; Ca guides treatment |
| Serum cortisol / ACTH stimulation test | Adrenal insufficiency |
| Plasma renin activity (PRA) + aldosterone | Hypoaldosteronism workup |
| Urine K⁺, Na⁺, creatinine | Calculate TTKG (transtubular K⁺ gradient) |
| Urine K⁺:Cr ratio | <13 mEq/g → inadequate renal K⁺ excretion |
| CBC | Haemolysis, leucocytosis/thrombocytosis (pseudohyperkalemia) |
| CK, urine myoglobin | Rhabdomyolysis |
| LDH, uric acid, phosphate | Tumour lysis syndrome |
| Drug review | ACEi, ARB, K-sparing diuretics, NSAIDs, heparin |
Transtubular K⁺ Gradient (TTKG):
- Normal >8–10 (adequate aldosterone-driven excretion)
- <5 in hyperkalemia → renal/aldosterone cause confirmed
Management
Three Mechanisms of Treatment:
| Mechanism | Drugs | Onset | Duration |
|---|---|---|---|
| 1. Membrane stabilisation (counteract cardiac toxicity) | Calcium | Seconds–minutes | 30–60 min |
| 2. Intracellular shift (drive K⁺ into cells) | Insulin+glucose, bicarbonate, β₂-agonists | Minutes | 2–6 hrs |
| 3. K⁺ removal from body | Diuretics, cation exchangers, dialysis | Hours | Definitive |
STEP 1 — Membrane Stabilisation (If ECG changes present)
IV Calcium — does NOT lower K⁺, but antagonises cardiac toxicity:
| Agent | Dose | Notes |
|---|---|---|
| Calcium gluconate 10% (preferred) | 10 mL (1 g) IV over 2–5 min; repeat in 5 min if ECG unchanged | Safer peripherally; 3× less elemental Ca than CaCl₂ |
| Calcium chloride 10% | 5–10 mL IV over 5–10 min | Central line only (vesicant); 3× more elemental Ca; used in cardiac arrest |
- Onset: 1–3 minutes; duration: 30–60 minutes — a temporising bridge only
- Caution: Do NOT give calcium in digoxin toxicity (hypercalcaemia + digoxin → "stone heart" / asystole)
- Repeat every 5 minutes if ECG changes persist
STEP 2 — Intracellular Shift (Lower plasma K⁺ temporarily)
A. Insulin + Glucose (most reliable)
- Regular insulin 10 units IV + 50 mL 50% dextrose (25g glucose) IV
- Onset: 15–30 min; effect lasts 4–6 hours; lowers K⁺ by ~0.6–1.0 mEq/L
- Monitor blood glucose every 30 minutes (hypoglycaemia risk)
- If euglycaemic/hyperglycaemic, give insulin without glucose
B. Sodium Bicarbonate
- 50–100 mEq (1–2 amps) IV over 5–10 minutes (1–2 mEq/kg)
- Most effective in metabolic acidosis (pH <7.3)
- Less effective in normal pH or ESRD (minimal benefit in isolation)
- Onset: minutes; lowers K⁺ by ~0.5–1.0 mEq/L
- Risk: hypernatraemia, volume overload, alkalosis
C. β₂-Agonists (Salbutamol/Albuterol)
- Nebulised salbutamol 10–20 mg (4× normal bronchodilator dose) over 10 min
- OR IV salbutamol 0.5 mg in 100 mL N/S over 15 min
- Onset: 30–60 min; lowers K⁺ by ~0.5–1.5 mEq/L
- Synergistic with insulin
- Caution: tachycardia, may worsen ischaemia; ~20–40% of patients resistant (especially on dialysis)
STEP 3 — Remove K⁺ from Body (Definitive)
A. Loop Diuretics (if renal function intact)
- Furosemide 40–80 mg IV (or 0.5–1 mg/kg in children)
- Enhances renal K⁺ excretion
- Only effective if urine output maintained
B. Cation Exchange Resins
| Agent | Mechanism | Dose | Route | Notes |
|---|---|---|---|---|
| Sodium polystyrene sulfonate (SPS / Kayexalate) | Exchanges Na⁺ for K⁺ in gut | 15–60 g orally or 30–60 g rectally | PO/rectal | Onset: 1–2 h (oral), <30 min (rectal); lowers K⁺ ~0.5–1 mEq/L; risk of intestinal necrosis (avoid with sorbitol in post-op) |
| Patiromer (Veltassa) | Binds K⁺ in GI tract | 8.4 g orally once daily | PO | Newer; no sorbitol; slower onset (7 hrs); chronic management |
| Sodium zirconium cyclosilicate (ZS-9, Lokelma) | K⁺ trap | 10 g TID × 48 hrs, then 5–10 g daily | PO | Fast onset (~1 hr); preferred newer agent; avoid in ileus |
C. Dialysis (most rapid, definitive K⁺ removal)
Indications:
- Serum K⁺ >6.5 mEq/L with anuric/oliguric renal failure
- Refractory hyperkalemia not responding to medical treatment
- Severe ECG changes + renal failure
- Concomitant volume overload + hyperkalemia
Haemodialysis (preferred over peritoneal dialysis for speed):
- Removes 25–50 mEq K⁺/hr
- Dialysate K⁺ = 0 or 1 mEq/L for maximum removal
Summary: Stepwise Treatment Table
| Step | Agent | Dose | Onset | Duration | Effect |
|---|---|---|---|---|---|
| Stabilise | IV Calcium gluconate | 10 mL 10% IV over 2–5 min | 1–3 min | 30–60 min | Membrane protection (no K⁺ change) |
| Shift | Insulin + 50% dextrose | 10U + 25g IV | 15–30 min | 4–6 hr | ↓K⁺ 0.6–1.0 mEq/L |
| Shift | Sodium bicarbonate | 50–100 mEq IV | Minutes | 2–4 hr | ↓K⁺ 0.5–1.0 mEq/L (if acidotic) |
| Shift | Nebulised salbutamol | 10–20 mg neb | 30 min | 2–4 hr | ↓K⁺ 0.5–1.5 mEq/L |
| Remove | Furosemide | 40–80 mg IV | 30–60 min | — | ↑ Renal excretion |
| Remove | Kayexalate | 15–60 g PO/rectal | 1–2 hr | — | ↓K⁺ 0.5–1.0 mEq/L |
| Remove | Haemodialysis | — | Rapid | Definitive | ↓K⁺ 1–2 mEq/L/hr |
Management of Underlying Cause
- Stop/reduce offending drugs (ACEi, ARB, K-sparing diuretics, NSAIDs)
- Treat acidosis, AKI, DKA
- Fludrocortisone in hypoaldosteronism/Addison's
- Low-potassium diet (avoid bananas, oranges, potatoes, tomatoes)
- Treat constipation (impairs GI K⁺ excretion)
Special Situations
| Situation | Modification |
|---|---|
| Digoxin toxicity | Avoid calcium; use insulin/glucose, magnesium, dialysis |
| Renal failure (no urine output) | Dialysis is definitive; resins have limited role |
| DKA | Serum K⁺ may be falsely high; true total body K⁺ is depleted — start K⁺ replacement once K⁺ <5.5 mEq/L and urine output established |
| Neonates/Infants | Phlebotomy haemolysis most common cause; calcium gluconate preferred (central if CaCl₂ used); kayexalate 1 g/kg |
Sources: Harrison's Principles of Internal Medicine, 22nd Ed. | Tintinalli's Emergency Medicine, 9th Ed. | Miller's Anesthesia, 10th Ed. | Rosen's Emergency Medicine | Current Surgical Therapy, 14th Ed.
Vitamin d deficiency op ghai paediatrics short note
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
I have comprehensive content. Now let me compile a thorough short note per OP Ghai framework.
Vitamin D Deficiency (Nutritional Rickets)
Short Note — OP Ghai Essential Pediatrics
Vitamin D Metabolism
Sunlight (UV 290–320 nm)
↓
7-dehydrocholesterol (skin)
↓
Previtamin D3 → Cholecalciferol (Vitamin D3)
↓ Liver (25-hydroxylase)
25-hydroxyvitamin D3 [25(OH)D3] — storage form; measured in serum
↓ Kidney (1α-hydroxylase — stimulated by PTH, low PO₄)
1,25-dihydroxyvitamin D3 [Calcitriol] — ACTIVE FORM
↓
Actions: ↑ Ca absorption (gut), ↑ Ca & PO₄ reabsorption (kidney),
↑ bone mineralisation, ↑ bone resorption (with PTH)
Dietary sources: Fortified milk, fish oil, salmon, sardines, cod liver oil, egg yolk, fortified cereals
Normal serum 25(OH)D levels:
| Status | Level |
|---|---|
| Sufficient | >30 ng/mL (>75 nmol/L) |
| Insufficient | 20–30 ng/mL |
| Deficient | <20 ng/mL (<50 nmol/L) |
| Severe deficiency | <10 ng/mL |
Definition
Nutritional Rickets = impaired mineralisation of growing bone (osteoid) at the growth plate due to vitamin D deficiency, calcium deficiency, or phosphate deficiency, occurring in the growing child.
Aetiology / Risk Factors
Decreased Production / Intake:
- Exclusive breastfeeding without supplementation (breast milk is low in vitamin D — most important risk factor)
- Dark skin (melanin reduces UV-mediated synthesis)
- Limited sunlight exposure — purdah, indoor confinement, high-latitude countries
- Inadequate dietary intake (vegetarian diet, no fortified foods)
Decreased Absorption:
- Coeliac disease, inflammatory bowel disease, cystic fibrosis, cholestatic liver disease
- Short bowel syndrome
Increased Requirement:
- Rapid growth phases — infancy, adolescence
- Prematurity (limited placental transfer; liver immaturity)
- Twin pregnancies
Maternal Deficiency:
- Maternal vitamin D deficiency during pregnancy → neonatal rickets
- Maternal dark skin, purdah, poor diet
Drugs:
- Anticonvulsants (phenobarbitone, phenytoin) — accelerate hepatic 25-hydroxylation → increased catabolism
- Rifampicin, glucocorticoids
Pathophysiology
↓ Vitamin D
↓
↓ Calcium absorption from gut
↓
Hypocalcaemia → ↑ PTH (Secondary hyperparathyroidism)
↓
PTH effects:
• ↑ Bone resorption → releases Ca but causes bone loss
• ↑ Renal Ca reabsorption
• ↑ Renal PO₄ excretion → HYPOPHOSPHATAEMIA
• ↑ Renal 1α-hydroxylase activity
Net result:
Ca may normalise (or be low in early/severe deficiency)
PO₄ = LOW (phosphaturia from high PTH)
ALP = HIGH (osteoblast activation)
↓ Ca × PO₄ product → failure of bone mineralisation
↓
Accumulation of unmineralised osteoid at growth plates → RICKETS
Clinical Features
Age of Presentation:
- 6 months to 2 years (most common; coincides with peak growth velocity and limited sun exposure in infancy)
- Neonatal rickets — in severely deficient mothers/preterm infants
A. Skeletal Features (most characteristic):
Skull:
- Craniotabes — softening of skull bones (occipital/parietal); "ping-pong ball" feel on pressing; earliest sign in infants
- Frontal bossing — prominence of frontal and parietal bones (caput quadratum)
- Delayed fontanelle closure
- Delayed dentition; enamel hypoplasia
Chest:
- Rachitic rosary — bead-like swellings at costochondral junctions (anterior ribs); a classic sign
- Harrison's sulcus (groove) — horizontal groove along lower chest wall at diaphragm insertion (due to softened rib pulling inward)
- Pigeon chest / pectus carinatum
Spine:
- Kyphosis, scoliosis, lordosis
Limbs:
- Widening of wrists and ankles (most reliable clinical sign after infancy) — expanded metaphyses
- Bow legs (genu varum) — in toddlers who bear weight
- Knock knees (genu valgum) — in older children
- Coxa vara (deformity of femoral neck)
- Greenstick fractures (pathological)
- Delayed walking
B. Non-Skeletal Features:
- Hypotonia ("floppy baby") — generalised muscle weakness
- Hypocalcaemic tetany — carpopedal spasm, laryngospasm, Chvostek's sign, Trousseau's sign
- Hypocalcaemic seizures (early infantile rickets, before skeletal signs develop)
- Pot belly (muscle hypotonia + hepatosplenomegaly)
- Dilated cardiomyopathy (rare but potentially fatal — responsive to Vit D treatment)
- Growth retardation
- Increased susceptibility to infections (impaired immunity)
- Dental: delayed eruption, enamel hypoplasia, caries
Investigations
Biochemistry:
| Test | Early Rickets | Established Rickets |
|---|---|---|
| Serum Ca | Normal (PTH compensates) | Low (if severe) |
| Serum PO₄ | Low | Low |
| Alkaline phosphatase (ALP) | ↑↑ (most sensitive marker) | ↑↑↑ |
| 25(OH)D (serum) | Low (<20 ng/mL) | Very low |
| 1,25(OH)₂D | Low or low-normal | Low |
| PTH | ↑ (secondary hyperparathyroidism) | ↑↑ |
| Urinary Ca | Low (hypocalciuria) | Low |
| Urinary PO₄ | High (phosphaturia from PTH) | High |
ALP elevation is the earliest and most sensitive biochemical marker Serum 25(OH)D is the best measure of vitamin D status
Radiology (Wrist X-ray — most informative):
Early:
- Widening and blurring of the growth plate (epiphyseal plate)
- Loss of zone of provisional calcification
Established:
- Cupping, fraying, and splaying of metaphysis (hallmark)
- Widened metaphysis
- Decreased bone density (osteopenia)
- Cortical spurs, stippling
Late (after treatment):
- Looser's zones (pseudofractures) in severe cases
- Reappearance of zone of provisional calcification (first sign of healing)
Classification / Types of Rickets
| Type | Cause | 25(OH)D | PTH | PO₄ | Ca |
|---|---|---|---|---|---|
| Nutritional VDD | Dietary/sun deficiency | ↓↓ | ↑ | ↓ | ↓/N |
| Calcium-deficiency rickets | Low Ca diet (Africa) | N | ↑ | N | ↓ |
| VD-dependent Type I (VDDR-I) | ↓ 1α-hydroxylase (AR) | N↑ | ↑ | ↓ | ↓ |
| VD-dependent Type II (VDDR-II) | VDR mutations; alopecia | ↑↑ | ↑ | ↓ | ↓ |
| X-linked hypophosphataemic rickets (XLH) | FGF23 excess (PHEX mutation) | N | N | ↓↓ | N |
| Renal osteodystrophy | CKD | Low | ↑ | ↑ | ↓ |
| Hepatic rickets | Liver disease | ↓ | ↑ | ↓ | ↓ |
Treatment
1. Vitamin D Supplementation
Daily therapy (preferred):
| Age | Daily Dose |
|---|---|
| Infants (<1 year) | 1000–2000 IU/day (25–50 mcg/day) for 3 months |
| Children 1–12 years | 3000–6000 IU/day for 3 months |
| Adolescents | 6000 IU/day for 3 months |
Stoss therapy (single/intermittent high-dose):
- Used when compliance is doubtful
- Oral or IM: 1,00,000–6,00,000 IU (100,000–600,000 IU) as a single dose or divided over 1–5 days
- Preferred in developing countries and resource-limited settings
- 3,00,000 IU (3 lakh IU) single oral dose — widely used in India (OP Ghai)
Maintenance after treatment: 400 IU/day
2. Calcium Supplementation
- Essential — especially in calcium-deficiency rickets
- Elemental calcium: 500–1000 mg/day orally
- Calcium gluconate or carbonate
- Given alongside vitamin D — prevents hypocalcaemia worsening post-treatment ("hungry bones")
3. Diet and Sun Exposure
- Encourage 15–30 minutes of sunlight daily (arms and face)
- Calcium-rich diet: milk, dairy, green leafy vegetables
- Fortified foods
4. Treatment of Complications
- Hypocalcaemic seizures/tetany: IV calcium gluconate 10% — 1–2 mL/kg slowly (0.5 mL/min) under ECG monitoring, then maintenance oral calcium
- Orthopedic correction: most deformities correct spontaneously after 2–3 years of treatment; persistent severe bowing after age 3–4 years may need surgery
5. Special Types
- VDDR Type I: 1-alpha-hydroxyvitamin D (Alfacalcidol) or Calcitriol
- VDDR Type II: Very high doses of calcitriol + calcium infusions
- XLH rickets: Phosphate supplements + calcitriol; burosumab (anti-FGF23) now available
- Hepatic rickets: Water-soluble vitamin D or TPGS formulations
Radiological Signs of Healing
- Reappearance of zone of provisional calcification — first sign (within 2–4 weeks)
- Reduction in metaphyseal cupping and fraying
- Improvement in bone density
- Normalisation of ALP (lags behind radiological healing — takes months)
Prevention
| Measure | Details |
|---|---|
| Routine supplementation | All breastfed infants: 400 IU/day from day 1 of life |
| Non-breastfed infants | 400 IU/day if intake <500 mL/day fortified formula |
| Pregnant women | 600–1000 IU/day (prevents neonatal rickets) |
| Sunlight | 15–30 min/day; arms and face |
| Fortified foods | Fortified milk, cereals, formula |
| High-risk groups | Dark skin, exclusive breastfeeding, purdah, prematurity |
Key Points for Exam
- Most common cause: Exclusive breastfeeding without VD supplementation in dark-skinned infants with limited sun exposure
- Earliest sign: Craniotabes (infants); widened wrists (older children)
- Most sensitive biochemical marker: ↑ Alkaline phosphatase
- Best serum marker for VD status: 25(OH)D (NOT 1,25(OH)₂D)
- Classic X-ray findings: Cupping, fraying, splaying of metaphysis
- Rachitic rosary = costochondral beading (rickets); not same as "rosary beads" of scurvy (subperiosteal haemorrhage)
- Stoss dose used in India: 3 lakh (300,000) IU single oral dose
- Harrison's sulcus = groove at lower chest margin (diaphragm insertion on soft ribs)
- Chvostek's sign = tapping facial nerve → facial twitch (hypocalcaemia)
- Trousseau's sign = BP cuff inflation above systolic → carpopedal spasm (hypocalcaemia)
Sources: Fitzpatrick's Dermatology, 9th Ed. | Harrison's Principles of Internal Medicine, 22nd Ed. — compiled per OP Ghai Essential Pediatrics framework.
This is a shared conversation. Sign in to Orris to start your own chat.