Tell me about retinitis pigmentosa
Reading File
Finding Sources
Searching PubMed
"retinitis pigmentosa"[MeSH Terms]
Searching the Web
retinitis pigmentosa treatment 2025 gene therapy voretigene
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have very comprehensive information. Let me compile a thorough response.
Retinitis Pigmentosa (RP)
Definition
Retinitis pigmentosa (RP) is not a single disease but a clinically and genetically diverse group of inherited diffuse retinal degenerations that primarily affect rod photoreceptors first, followed by progressive cone and retinal pigment epithelium (RPE) degeneration. Despite the name, it is not an inflammatory condition - the term "retinitis" is a historical misnomer.
- Kanski's Clinical Ophthalmology, 10th ed.
- Harrison's Principles of Internal Medicine 22E
Epidemiology
- Most common hereditary retinal degeneration
- Prevalence: 1 in 3,000 to 1 in 5,000 people worldwide
- Approximately 1.5 million people affected globally
- Age of onset, rate of progression, and severity correlate with the mode of inheritance
Genetics & Inheritance
RP can arise sporadically or be inherited. Inheritance patterns:
| Pattern | Notes |
|---|---|
| Autosomal dominant (AD) | Best prognosis; often due to mutations in the rhodopsin (RHO) gene |
| Autosomal recessive (AR) | Intermediate severity; associated with USH2A, EYS gene mutations |
| X-linked | Most severe; ~90% due to RPGR gene mutations; causes central vision loss to 6/60 or worse by the fifth decade |
Over 100 gene loci are implicated in non-syndromic RP. Key gene categories affected:
- Phototransduction cascade (e.g., rhodopsin)
- Retinoid cycle
- Photoreceptor structure (e.g., peripherin)
- RPE function
Despite this, about half of affected individuals still have no identified molecular genetic abnormality.
Pathophysiology
Rod photoreceptors degenerate first, with cones dying secondarily. Cell death occurs by apoptosis; cone death is partly mediated through RIP kinase activation. As photoreceptors die:
- Retinal vessels constrict
- Optic nerve head atrophies ("waxy pallor")
- Pigment from dying RPE cells accumulates around blood vessels, forming the classic "bone spicules"
Clinical Features
Symptoms (roughly in order of progression):
- Nyctalopia (night blindness) - usually the first symptom; caused by rod degeneration
- Peripheral visual field loss - mid-peripheral ring scotomas that coalesce
- Photopsia (flashing lights) - not uncommon
- Reduced central acuity - later feature, unless cataract develops early
Classic triad on fundoscopy:
- Bone-spicule pigmentation - irregular black pigment deposits in the mid-peripheral retina
- Arteriolar attenuation - narrowing of retinal vessels
- "Waxy" disc pallor - optic nerve head atrophy
Here is the classic fundus appearance:
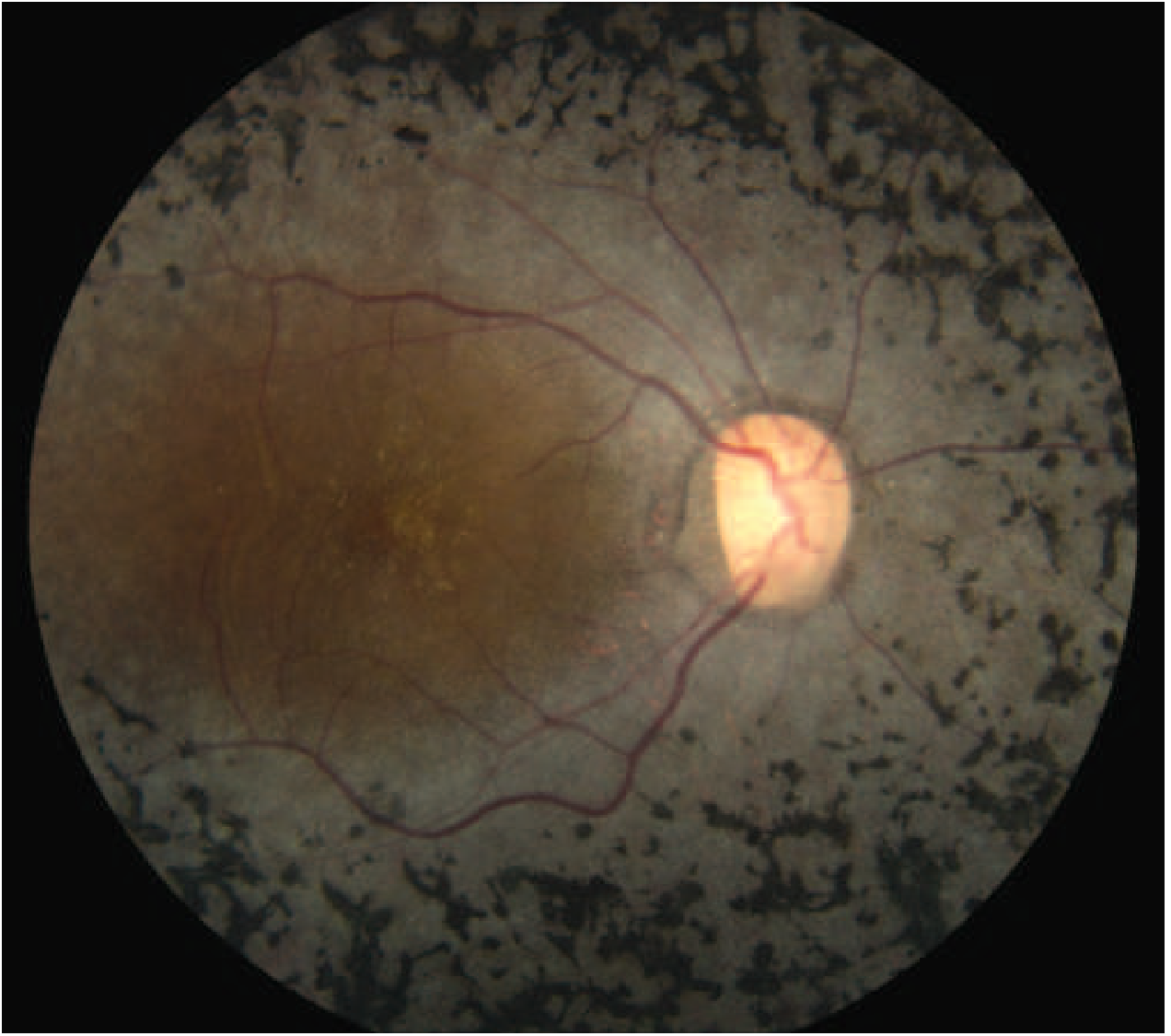
Fundus photo showing characteristic bone-spicule pigmentation scattered throughout the retina with macular sparing of central vision - Harrison's Principles of Internal Medicine 22E, Fig. 34-16
Other ocular findings:
- Myopia (common)
- Cystoid macular oedema (CMO) in ~15%
- Macular atrophy (~40%)
- Epiretinal membrane (ERM)
- Posterior subcapsular cataract (common complication in all forms)
- Open-angle glaucoma (~3%)
- Optic disc drusen (more frequent than general population)
Investigations
| Test | Findings |
|---|---|
| Full-field ERG | Most sensitive diagnostic test; early: reduced scotopic (rod) responses; late: extinguished |
| Visual field perimetry | Mid-peripheral scotomas coalescing to "tunnel vision" |
| Fundus autofluorescence (FAF) | Abnormal perimacular ring of hyperautofluorescence; patchy hypo-AF in periphery; distinguishes RP from normal in 95% of cases |
| OCT | Detects macular oedema and atrophy; prognostic value |
| Dark adaptation test | Prolonged; useful in early/equivocal cases |
| Genetic analysis | Identifies specific mutations for counselling and clinical trial eligibility |
Syndromic Forms
In 20-30% of cases, RP is associated with a systemic disorder. Key syndromes include:
| Syndrome | Features |
|---|---|
| Usher syndrome | RP + sensorineural hearing loss (most common) |
| Bardet-Biedl syndrome | RP + obesity, polydactyly, renal anomalies, cognitive impairment |
| Refsum disease | RP + peripheral neuropathy, ataxia, deafness (phytanic acid accumulation) |
| Bassen-Kornzweig (abetalipoproteinemia) | RP + ataxia, acanthocytosis |
| Kearns-Sayre syndrome | RP + external ophthalmoplegia, cardiac conduction defects (mitochondrial) |
| Leber congenital amaurosis (LCA) | Severe early-onset form (~5% of RP); target for gene therapy |
| NARP syndrome | Neuropathy, Ataxia, Retinitis Pigmentosa (mitochondrial) |
RP Mimics (Important Differentials)
Several conditions can produce a fundus appearance resembling RP:
- Syphilis (always investigate and exclude)
- Chloroquine / hydroxychloroquine toxicity - causes bull's-eye maculopathy; requires regular monitoring with OCT
- Thioridazine (phenothiazine) toxicity
- Trauma, old retinal detachment
Prognosis
| Inheritance | Prognosis |
|---|---|
| Autosomal dominant | Best - slower progression |
| Autosomal recessive | Intermediate |
| X-linked | Worst - central vision often reduced to 6/60 or less by age 50 |
Management
Currently no curative treatment is commercially available for most forms of RP, but management includes:
Supportive:
- Low vision aids, tinted lenses (reduce photophobia)
- Avoidance of high-intensity light
- Treatment of complications (e.g., cataract surgery, carbonic anhydrase inhibitors for CMO)
- Vitamin A palmitate supplementation (some evidence for slowing progression; must be monitored for toxicity)
Gene Therapy (the most active area):
- Luxturna (voretigene neparvovec-rzyl) - the first FDA-approved gene therapy for an inherited retinal disease; treats RPE65-mutation-associated retinal dystrophy (LCA2), which represents only ~0.3-1% of RP cases. Delivered as subretinal injection.
- RPGR-targeted therapies for X-linked RP (most common X-linked form) are in active trials. A 2026 systematic review and meta-analysis (PMID 41773775) evaluated gene therapy for RPGR-associated XLRP - this is a rapidly evolving area.
- MCO-010 (optogenetic gene therapy) - delivers channelrhodopsin proteins to surviving retinal cells, potentially restoring some light sensitivity even in advanced RP regardless of mutation; up to 50% of patients gained measurable vision improvement in early trials (AAO 2024 data).
- Botaretigene sparoparvovec (bota-vec) and laru-zova are additional XLRP gene therapy candidates in Phase 3 trials.
Artificial retina / retinal prostheses (e.g., Argus II) - for end-stage RP.
Genetic counselling is recommended for all patients and families.
Female Carriers (X-linked RP)
Female carriers may show:
- Normal fundi, OR
- A golden-metallic "tapetal" reflex at the macula
- Small peripheral bone-spicule pigmentation
- Centrifugal hyperautofluorescent lines on FAF imaging (most distinctive carrier sign)
Sources: Kanski's Clinical Ophthalmology 10th ed., Harrison's Principles of Internal Medicine 22E, Robbins, Cotran & Kumar Pathologic Basis of Disease
Recent evidence update: A 2026 meta-analysis on RPGR gene therapy for X-linked RP (PMID 41773775) and an AI-based diagnostic accuracy review (PMID 40719545) reflect very active ongoing progress. Phase 3 trial results (LUMEOS study) for bota-vec showed directionally supportive but not statistically significant improvement at 52 weeks, indicating gene therapy for RP remains a work in progress beyond the established RPE65 indication.
This is a shared conversation. Sign in to Orris to start your own chat.