Give me a pathology-based summary of the topic von willebrands disease. It should explain different, the required mechanisms so that by hearting can be prevented. And it should cover almost 100% of topics that can be asked for NEET PG clinical questions from pathology. It should be an easily revisable format.
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content needed from authoritative pathology sources. Let me compile the comprehensive NEET PG-focused summary:
Von Willebrand Disease (vWD) — NEET PG Pathology Master Summary
Source refs: Robbins & Kumar Basic Pathology, p. 430 | Goldman-Cecil Medicine | Henry's Clinical Diagnosis & Management by Laboratory Methods
1. THE BASICS (One-liner anchors)
| Feature | Detail |
|---|---|
| Most common | Inherited bleeding disorder overall |
| Gene | VWF gene — 52 exons, chromosome 12 |
| Inheritance | Autosomal dominant (most types) |
| Exception | Type 3 → autosomal recessive |
| Prevalence | ~1% of population (significant bleeding: 1:1000) |
2. VWF — STRUCTURE & DUAL FUNCTION (The Core Mechanism)
vWF is a large multimeric glycoprotein synthesized in:
- Endothelial cells → stored in Weibel-Palade bodies (main source)
- Megakaryocytes → stored in α-granules of platelets (10–20% of total)
Two essential roles:
Role 1 — Platelet Adhesion (Primary Hemostasis)
Vascular injury → Subendothelial collagen exposed
↓
vWF (A3 domain) binds collagen types I & III
vWF (A1 domain) binds collagen types IV & VI
↓
vWF bridge: Collagen ←→ vWF ←→ Platelet GpIb receptor
↓
Platelet adhesion → activation → GpIIb/IIIa exposed
↓
Fibrinogen bridges GpIIb/IIIa → platelet aggregation
High-molecular-weight (HMW) multimers are the most functionally active — they bind collagen and platelets with highest affinity, especially under high-shear conditions.
Role 2 — Carrier/Chaperone for Factor VIII (Secondary Hemostasis)
vWF + Factor VIII → noncovalent complex in plasma
↓
Protects Factor VIII from premature proteolytic degradation & clearance
↓
On platelet activation: Factor VIII dissociates from vWF
↓
Factor VIII + activated Factor IX → activates Factor X (intrinsic pathway)
ADAMTS13 — The Regulator
- A specific metalloprotease (disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13)
- Cleaves ultra-large vWF multimers → keeps multimer size in check
- ADAMTS13 deficiency → ultra-large multimers persist → spontaneous platelet aggregation → TTP (contrast with vWD!)
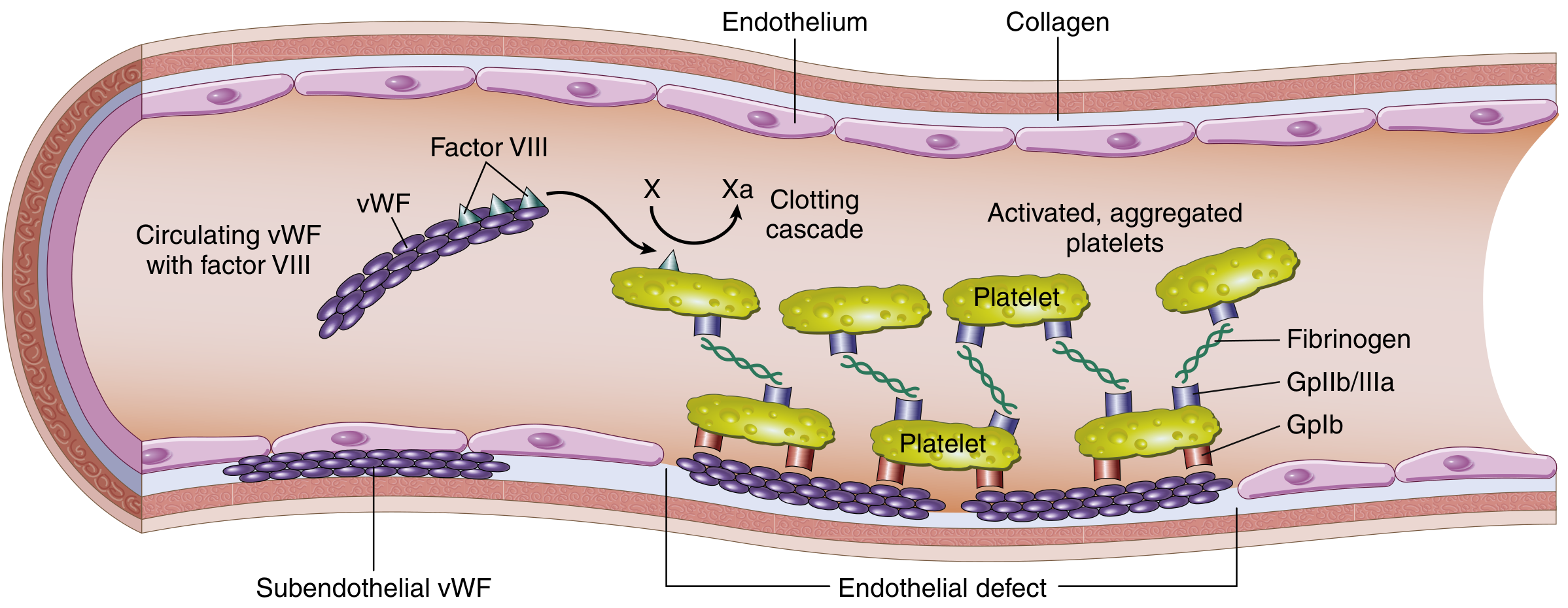
Fig: Factor VIII–vWF complex. vWF in subendothelial matrix mediates platelet adhesion (GpIb) and aggregation (GpIIb/IIIa via fibrinogen). — Robbins & Kumar, p. 430
3. CLASSIFICATION OF vWD — The Master Table
| Type | Defect | Multimers | Inheritance | Key Distinguishing Feature |
|---|---|---|---|---|
| 1 (most common ~75%) | ↓ Quantity (partial) | All sizes present, just reduced | AD | Mild–moderate bleeding; vWF antigen & activity both ↓ proportionally |
| 1C | ↑ Clearance of vWF | All sizes, rapid clearance | AD | vWFpp/vWF:Ag ratio elevated; DDAVP does NOT work |
| 2A | Loss of HMW multimers — not synthesized | HMW multimers absent | AD | ↓ ristocetin-induced platelet aggregation (RIPA); loss-of-function |
| 2B | Abnormal "hyperfunctional" HMW multimers → rapidly cleared | HMW multimers absent (but gain-of-function mutation) | AD | Spontaneous platelet aggregation; mild thrombocytopenia; DDAVP contraindicated |
| 2M | ↓ Platelet binding (A1 domain mutation) | Normal multimer pattern | AD | ↓ RIPA; multimers normal on gel |
| 2N | ↓ Factor VIII binding (D′/D3 domain) | Normal | AR | Resembles hemophilia A (↓ FVIII); RIPA normal |
| 3 (most severe) | Total absence of vWF | Absent | AR | ↓↓ FVIII → hemophilia-like + severe mucosal bleeding; bleeds from both primary & secondary hemostasis |
Mnemonic for Type 2: 2A = Absent HMW (not synthesized); 2B = Bad HMW (hyperfunctional, consumed); 2M = Malformed binding site; 2N = No FVIII binding
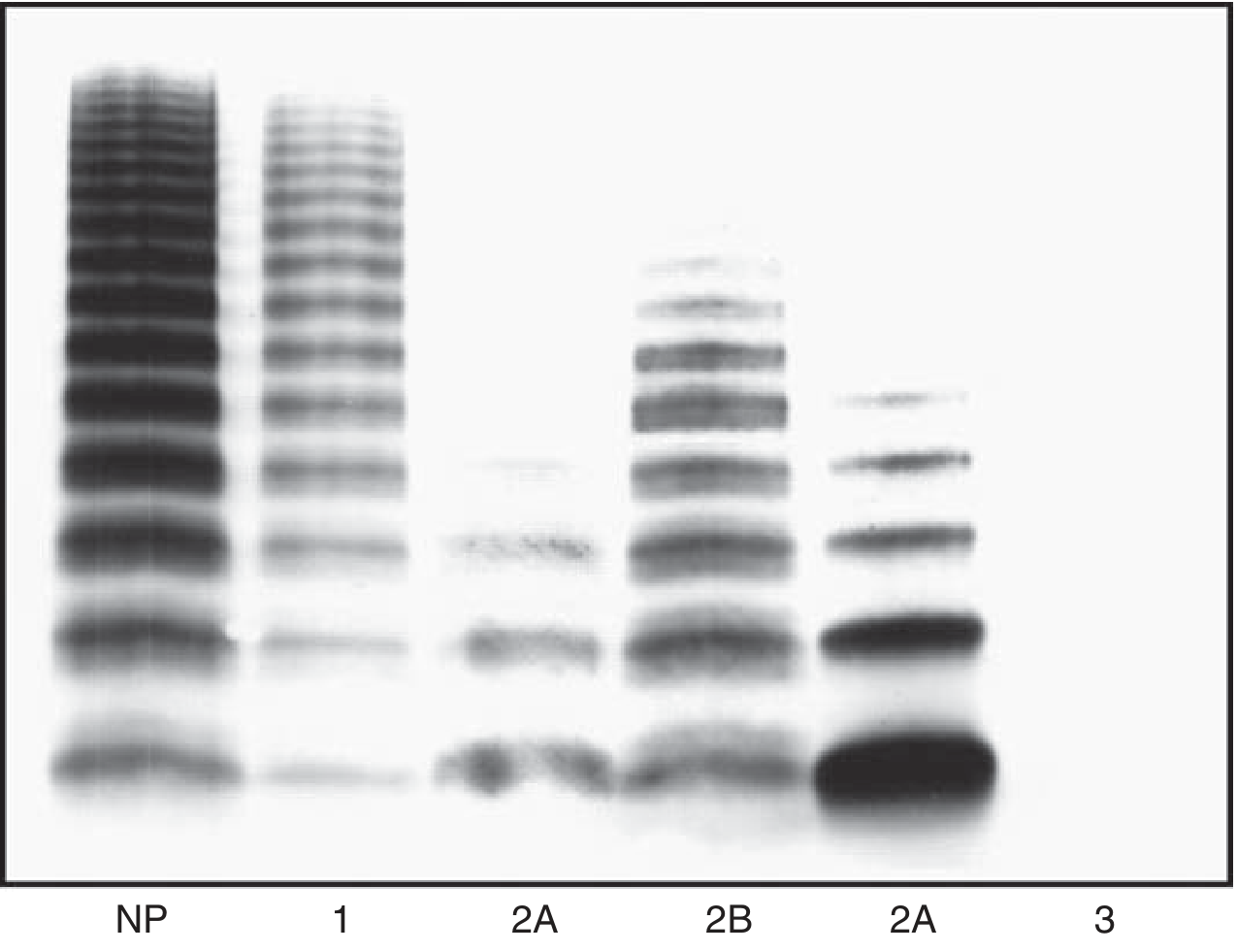
Fig: SDS-agarose gel of vWF multimers. Type 1 = reduced all bands; Type 2A/2B = loss of upper (HMW) bands; Type 3 = essentially absent. — Henry's Clinical Diagnosis, p. 979
4. CLINICAL FEATURES
Mucocutaneous bleeding pattern (contrast: hemophilia → deep tissue/hemarthrosis)
- Epistaxis (most common)
- Gingival bleeding
- Menorrhagia (often the presenting symptom in females)
- Prolonged bleeding from minor cuts/wounds
- Easy bruising
- GI bleeding
- Post-surgical / dental extraction bleeding
- Type 3 / severe vWD only: hemarthrosis, deep muscle hematomas (due to ↓↓ FVIII)
Key contrast with hemophilia: vWD → mucosal/surface bleeding | Hemophilia → deep tissue bleeding
5. LABORATORY DIAGNOSIS
Screening Tests
| Test | Result in vWD |
|---|---|
| Bleeding time (BT) / PFA-100 closure time | Prolonged (platelet plug defect) |
| aPTT | Normal (Type 1, 2A, 2B, 2M) / Prolonged (Type 2N, Type 3 — due to ↓ FVIII) |
| PT | Normal |
| Platelet count | Usually normal; mild thrombocytopenia in Type 2B |
Specific Diagnostic Tests
| Test | What it Measures | Findings |
|---|---|---|
| vWF antigen (vWF:Ag) | Total vWF protein level | ↓ in Type 1, 3; normal in 2M, 2N |
| vWF ristocetin cofactor activity (vWF:RCo) | Functional activity (platelet binding) | ↓ in most types |
| Ristocetin-induced platelet aggregation (RIPA) | High-dose (1.2 mg/mL) = platelet adhesion; Low-dose (0.5 mg/mL) = spontaneous aggregation test | ↓ RIPA (high-dose) in 2A, 2M; ↑ RIPA (low-dose) in 2B |
| Factor VIII activity | Intrinsic pathway | ↓ in Type 2N, Type 3; mildly ↓ in Type 1 |
| vWF multimer analysis (SDS-agarose gel) | Multimer size distribution | Absent HMW multimers in 2A, 2B; absent all in Type 3 |
| vWF:CBA (collagen binding activity) | vWF binding to collagen | ↓ in types with HMW loss; ratio vWF:CBA/vWF:Ag <0.6 suggests multimer loss |
| vWFpp/vWF:Ag ratio | vWF clearance rate | ↑ in Type 1C (increased clearance) |
ABO Blood Group Effect (Exam Trap!)
- Blood group O individuals have ~25% lower vWF levels than non-O
- Can push borderline cases into "abnormal" range
- Treatment decisions based on vWF level, NOT modified for ABO status
- vWF level 30–50 IU/dL → labeled "low vWF" rather than definitive Type 1 vWD
Diagnostic Criteria Summary
- Normal range: vWF 50–150 IU/dL
- vWF:RCo/vWF:Ag ratio ≥0.6 = normal function
- <0.6 = functional deficiency (suggests HMW multimer loss)
6. RISTOCETIN — The Exam Favorite
| Situation | Explanation |
|---|---|
| Ristocetin causes platelet agglutination in normal plasma | Ristocetin activates vWF to bind GpIb → platelet aggregation |
| RIPA ↓ in vWD (all types except 2B at low dose) | Not enough functional vWF to bridge platelet-collagen |
| RIPA ↑ at LOW dose in Type 2B | Gain-of-function mutation → HMW multimers bind GpIb spontaneously |
| RIPA normal in Type 2N | Platelet-binding domain intact; only FVIII binding domain defective |
| Bernard-Soulier syndrome | RIPA absent (no GpIb receptor on platelets) — contrast with vWD where receptor is normal |
7. PATHOGENESIS MECHANISMS — TYPE BY TYPE
Type 1
Quantitative ↓ in vWF production (all multimers reduced)
→ Less vWF available to bridge platelet-collagen
→ Impaired primary hemostasis
→ Mild ↓ FVIII (clinical significance minimal)
Type 2A
Mutation → failure to synthesize HMW multimers
→ Only low-MW forms circulate
→ Poor platelet bridging (HMW most active)
→ Functional deficiency despite some vWF present
Type 2B
Gain-of-function mutation in A1 domain
→ HMW multimers bind GpIb SPONTANEOUSLY (even without injury)
→ HMW multimers consumed → absent from plasma
→ Spontaneous platelet clumping → mild thrombocytopenia
→ (Mimics TTP mechanism but different etiology)
Type 2N (Normandy variant)
Mutation in D'/D3 domain of vWF (FVIII binding site)
→ vWF cannot carry FVIII → FVIII degraded prematurely
→ ↓ FVIII levels
→ Mimics Hemophilia A clinically
→ BUT: X-linked family history absent; females also affected equally
→ RIPA normal (platelet binding intact)
Type 3
Homozygous/compound heterozygous mutation
→ Complete absence of vWF
→ No platelet adhesion → severe mucosal bleeding
→ No FVIII carrier → FVIII level <1% → hemophilia-like hemarthrosis
→ Both pathways compromised
8. TREATMENT
| Agent | Mechanism | Indication | Contraindication |
|---|---|---|---|
| DDAVP (Desmopressin) | Releases stored vWF from Weibel-Palade bodies | Type 1 (first-line), Type 2A | Type 2B (causes more spontaneous aggregation, worsens thrombocytopenia); Type 1C (increased clearance negates effect); Type 3 (no stores to release) |
| vWF concentrates (plasma-derived or recombinant) | Replaces vWF directly | Type 2B, Type 3, Type 1C, DDAVP-unresponsive | — |
| Antifibrinolytics (tranexamic acid, ε-aminocaproic acid) | Prevent fibrinolysis | Adjunct for mucosal bleeding, dental procedures | — |
| Cryoprecipitate | Contains vWF + FVIII + fibrinogen | Where specific concentrates unavailable | Less preferred (risk of transfusion reactions) |
| Recombinant FVIII | Not vWF replacement | Type 2N, Type 3 (for FVIII deficiency component) | — |
DDAVP works via V2 receptors on endothelial cells → exocytosis of Weibel-Palade bodies → surge in vWF release
9. KEY COMPARISONS FOR MCQs
vWD vs Hemophilia A
| Feature | vWD | Hemophilia A |
|---|---|---|
| Defect | vWF (± secondary FVIII) | Factor VIII only |
| Gene | Chromosome 12 | X chromosome (Xq28) |
| Inheritance | AD (most) / AR (Type 3) | X-linked recessive |
| Gender | M = F affected | Mainly males |
| Bleeding type | Mucocutaneous | Deep tissue, hemarthrosis |
| BT/PFA | Prolonged | Normal |
| aPTT | Normal (usually) | Prolonged |
| RIPA | Decreased (except 2B low-dose) | Normal |
| Treatment | DDAVP / vWF concentrates | FVIII concentrates |
vWD vs Bernard-Soulier Syndrome (BSS)
| Feature | vWD | BSS |
|---|---|---|
| Defect | vWF (ligand) | GpIb (receptor) absent |
| RIPA | ↓ | Absent (no receptor) |
| RIPA corrected by normal plasma? | Yes (adds vWF) | No (receptor defect) |
| Platelet count | Normal (except 2B) | ↓ (giant platelets) |
| Platelet morphology | Normal | Large/giant platelets |
vWD vs TTP (ADAMTS13 link)
| Feature | vWD | TTP |
|---|---|---|
| ADAMTS13 | Normal | Absent/inhibited |
| vWF multimers | Reduced/absent | Ultra-large (not cleaved) |
| Effect | Bleeding | Thrombotic microangiopathy |
| Type 2B similarity | HMW loss + thrombocytopenia | Different mechanism |
10. HIGH-YIELD NEET PG EXAM POINTS
- Most common inherited bleeding disorder = vWD (not hemophilia)
- Type 1 = most common vWD subtype (75%)
- Type 3 = most severe (autosomal recessive, complete absence)
- HMW multimers are most active — their loss → functional deficiency
- DDAVP is contraindicated in Type 2B (worsens thrombocytopenia and bleeding)
- Type 2B = gain-of-function mutation → spontaneous platelet aggregation → thrombocytopenia + absent HMW multimers
- Type 2N mimics hemophilia A — but AD/AR, X-linked pattern absent
- RIPA ↑ at low-dose ristocetin = Type 2B signature
- RIPA used to differentiate: ↓ in vWD (platelet binding impaired) vs absent in Bernard-Soulier (no GpIb)
- vWF stored in Weibel-Palade bodies (endothelial) and α-granules (platelets)
- ADAMTS13 cleaves vWF — deficiency → TTP (not vWD)
- ABO blood group O → 25% lower vWF (can falsely suggest vWD)
- Chromosome 12 (VWF gene) — exam trap vs X-chromosome (Factor VIII gene itself)
- vWF carries FVIII — in Type 3/2N, ↓ FVIII → prolonged aPTT
- Bleeding time/PFA prolonged — primary hemostasis defect (platelet plug formation fails)
- Cryoprecipitate contains vWF + FVIII + fibrinogen + factor XIII + fibronectin
- Type 1C (increased clearance) — vWFpp/vWF:Ag ratio elevated; DDAVP ineffective
- vWF:RCo / vWF:Ag < 0.6 = qualitative defect (functional below antigen level)
- Menorrhagia is a common presenting symptom in females — vWD underdiagnosed in women
- Type 2A vs 2B on gel: both lack HMW bands, distinguished by RIPA at low dose (↑ in 2B only)
11. QUICK REVISION MNEMONIC GRID
vWD TYPE DEFECT MULTIMERS RIPA DDAVP FVIII
Type 1 ↓ Quantity All ↓ ↓ (high) YES Mildly ↓
Type 1C ↑ Clearance All ↓ (fast) ↓ NO Mildly ↓
Type 2A No HMW synthesis HMW absent ↓ Usually Normal
Type 2B HMW consumed HMW absent ↑ at low NO! Normal
Type 2M ↓ Platelet bind Normal ↓ Usually Normal
Type 2N ↓ FVIII binding Normal Normal NO ↓↓
Type 3 Total absence Absent Absent NO ↓↓↓
Sources: Robbins & Kumar Basic Pathology, p. 430 | Goldman-Cecil Medicine, Vol. 2 | Henry's Clinical Diagnosis and Management by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.