I now have comprehensive content. Here is a thorough synthesis on GABA and Glutamate — their roles in disease and pharmacology.
GABA & Glutamate: Role in Disease and Pharmacology
These two amino acid neurotransmitters are the primary brakes and accelerators of the CNS. Glutamate is the major excitatory neurotransmitter; GABA is the major inhibitory neurotransmitter. Their balance governs virtually all brain activity.
GLUTAMATE
Synthesis & Recycling
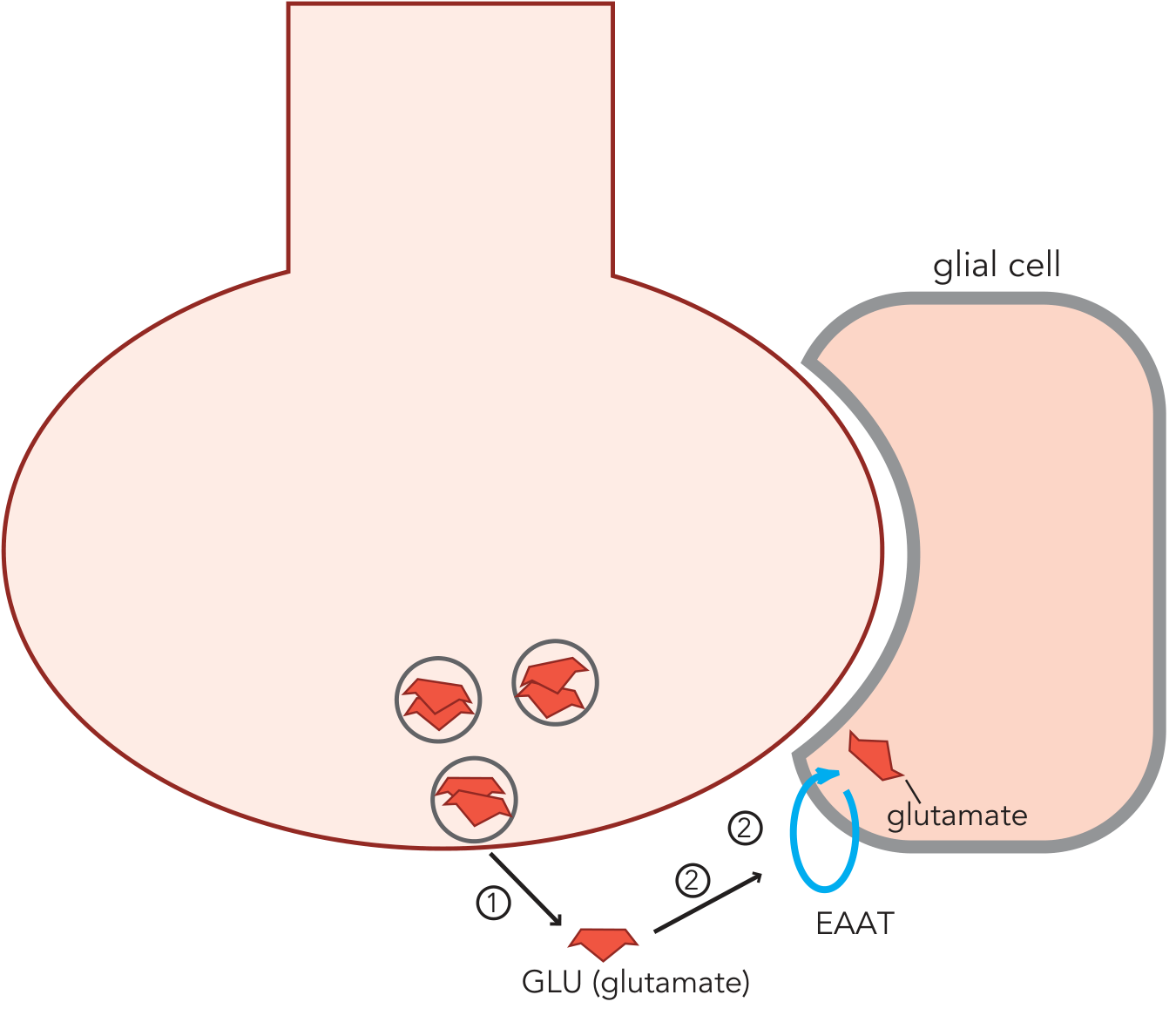
Glutamate is synthesized from glutamine (supplied by glial cells) via the enzyme glutaminase in neuronal mitochondria. After release, it is cleared from the synapse predominantly by excitatory amino acid transporters (EAATs) on glial cells, where it is converted back to glutamine by glutamine synthetase — completing the glutamate–glutamine cycle.
Glutamate released from the presynaptic terminal (1) is taken up into glial cells via EAAT (2) — Stahl's Essential Psychopharmacology
Receptors
Glutamate acts on two major receptor families:
| Receptor type | Subtypes | Ion / Mechanism | Notes |
|---|
| Ionotropic | AMPA | Na⁺, K⁺ | Fast excitatory transmission |
| Kainate | Na⁺, K⁺ | |
| NMDA | Na⁺, K⁺, Ca²⁺ | Requires both glutamate AND glycine co-agonist; blocked by Mg²⁺ at rest; critical for LTP and memory |
| Metabotropic | mGluR1–8 | G-protein-coupled | Modulatory roles; mGluR2/3 implicated in schizophrenia |
— Ganong's Review of Medical Physiology, Table 7-2
Glutamate in Disease
1. Schizophrenia — NMDA Receptor Hypofunction
The glutamate hypothesis of schizophrenia is grounded in the observation that NMDA receptor antagonists (ketamine, phencyclidine/PCP) produce a syndrome indistinguishable from schizophrenia — including positive, negative, and cognitive symptoms.
"Glutamate is the major excitatory amino acid neurotransmitter in the brain and is sometimes considered the 'master switch' of the brain... glutamate has attained a key theoretical role in the hypothesized pathophysiology of schizophrenia."
— Stahl's Essential Psychopharmacology
The mechanism: NMDA receptor hypofunction on GABAergic parvalbumin interneurons in the prefrontal cortex → disinhibition → downstream dopamine dysregulation (hyperdopaminergia in mesolimbic pathways → positive symptoms; hypodopaminergia in mesocortical pathways → negative/cognitive symptoms).
In schizophrenia, postmortem studies consistently show:
- Decreased hippocampal AMPA receptor subunit expression
- Downregulated NMDA GluN1 subunit in temporal cortex, hippocampus, and thalamus
- Decreased mGluR2 and mGluR3 expression
2. Excitotoxicity
Excess glutamate overwhelms EAAT clearance → sustained NMDA receptor activation → Ca²⁺ overload → mitochondrial dysfunction and neuronal death. This is implicated in stroke, traumatic brain injury, ALS, and Alzheimer's disease.
3. Depression
Glutamate dysfunction in the PFC and hippocampus contributes to depression. This is the basis for ketamine's rapid antidepressant effect (NMDA receptor blockade).
Glutamate Pharmacology
| Drug | Mechanism | Clinical Use |
|---|
| Memantine | Low-affinity NMDA antagonist | Alzheimer's disease (moderate–severe) |
| Ketamine | NMDA antagonist | Anesthesia; treatment-resistant depression (esketamine) |
| Riluzole | Inhibits glutamate release (blocks Na⁺ channels) | ALS — only disease-modifying drug |
| Perampanel | AMPA receptor antagonist | Epilepsy (focal seizures) |
| Lamotrigine | Inhibits voltage-gated Na⁺ channels → reduces glutamate release | Epilepsy, bipolar disorder |
GABA (γ-Aminobutyric Acid)
Synthesis & Termination
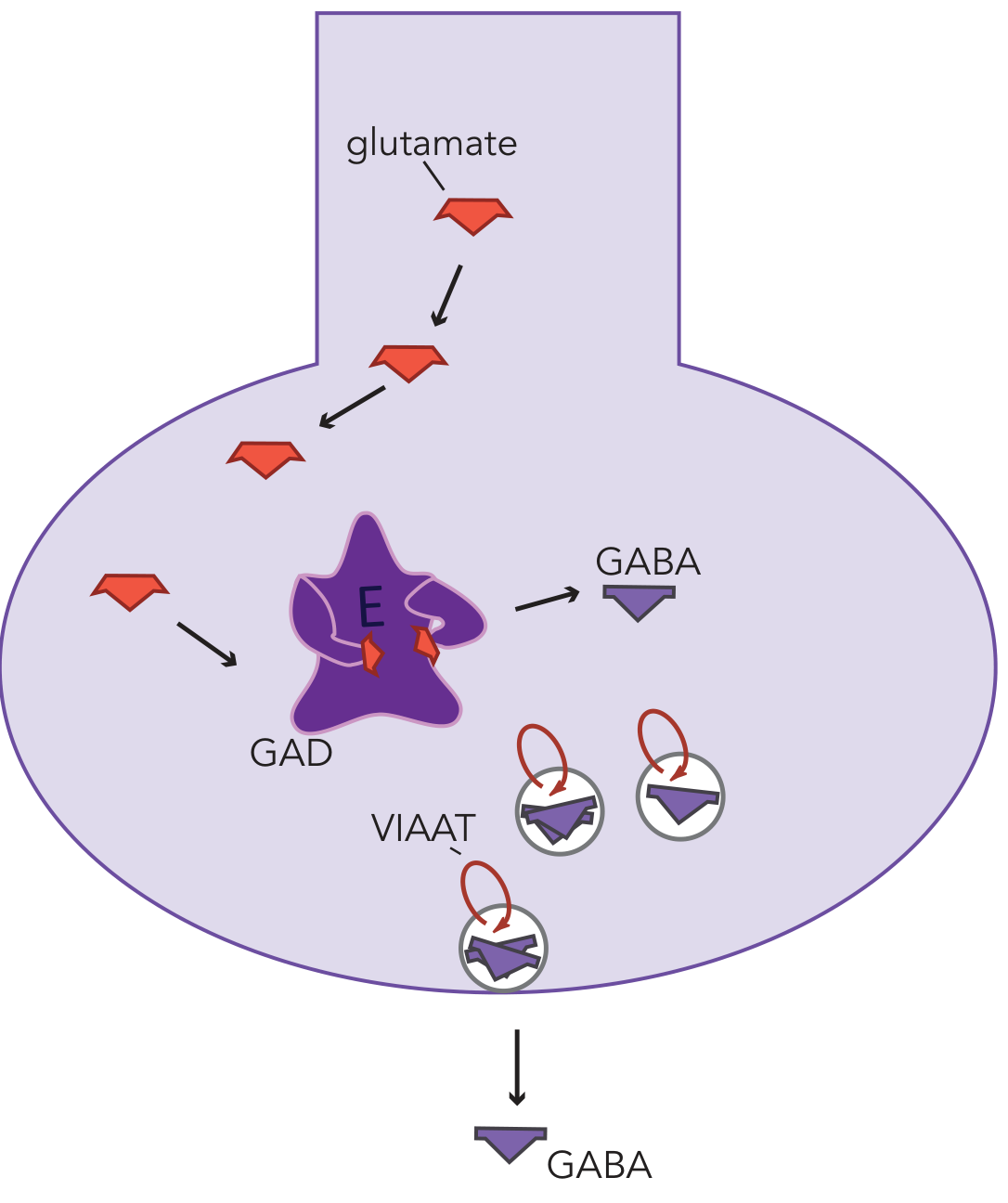
GABA is synthesized from glutamate by the enzyme glutamic acid decarboxylase (GAD) — requiring pyridoxal phosphate (vitamin B6) as cofactor. After synthesis, GABA is packaged into vesicles by vesicular inhibitory amino acid transporters (VIAATs) and released.
GABA action is terminated by:
- Reuptake via the GABA transporter (GAT) back into the presynaptic neuron
- Enzymatic degradation by GABA-transaminase (GABA-T)
GABA production: glutamate is converted to GABA by GAD, then stored in vesicles via VIAAT — Stahl's Essential Psychopharmacology
Receptors
| Receptor | Type | Mechanism | Key features |
|---|
| GABA-A | Ligand-gated Cl⁻ channel | ↑Cl⁻ influx → hyperpolarization | Site of benzodiazepines, barbiturates, alcohol, propofol, neurosteroids; subunit composition determines sensitivity |
| GABA-B | G-protein-coupled | Opens K⁺ channels, closes Ca²⁺ channels | Presynaptic autoreceptor; target of baclofen |
| GABA-C (ρ) | Ligand-gated Cl⁻ channel | Similar to GABA-A | Predominantly in retina |
GABA-A subunit diversity is pharmacologically important: receptors containing γ subunits are synaptic, mediate phasic inhibition, and are benzodiazepine-sensitive; those with δ subunits are extrasynaptic, mediate tonic inhibition, and are benzodiazepine-insensitive. — Stahl's Essential Psychopharmacology
GABA in Disease
1. Epilepsy
Reduced GABAergic inhibition → uncontrolled neuronal firing. This is the central mechanism of most seizure disorders. GAD mutations, GABA-A subunit mutations (GABRG2, GABRA1), and GABA transporter defects all cause epilepsy syndromes.
2. Anxiety Disorders
Reduced tonic GABA-A activity in amygdala, hippocampus, and cortex. "GABA is the most widespread inhibitory neurotransmitter in the brain. GABA-A receptors... mediate fast inhibitory postsynaptic potentials." — Neuroscience: Exploring the Brain
3. Schizophrenia
A consistent finding in postmortem PFC is reduced GAD67 mRNA in parvalbumin-positive interneurons — without loss of neurons themselves. This reduces GABA synthesis in PV+ interneurons, impairing inhibitory gating and contributing to cognitive dysfunction. GABA circuitry is regulated by schizophrenia-risk genes BDNF, DISC1, and NRG1. — Kaplan & Sadock's Comprehensive Textbook of Psychiatry
4. Alcohol Use Disorder
Alcohol potentiates GABA-A receptors and inhibits NMDA receptors. Chronic use leads to compensatory downregulation of GABA-A; abrupt withdrawal reverses this → neuronal hyperexcitability → seizures, delirium tremens.
5. Hepatic Encephalopathy
Ammonia → increased brain glutamine → astrocyte swelling + enhanced GABAergic tone → altered consciousness.
GABA Pharmacology
| Drug | Mechanism | Clinical Use |
|---|
| Benzodiazepines (diazepam, lorazepam) | Positive allosteric modulator at GABA-A (↑frequency of Cl⁻ channel opening) | Anxiety, acute seizures, alcohol withdrawal |
| Barbiturates (phenobarbital, thiopental) | Positive allosteric modulator at GABA-A (↑duration of Cl⁻ channel opening); at high doses, directly open channel | Epilepsy, anesthesia |
| Propofol | Allosterically ↑ GABA binding to GABA-A | IV anesthesia |
| Vigabatrin | Irreversible GABA-T inhibitor → ↑GABA levels | Infantile spasms, tuberous sclerosis |
| Tiagabine | GAT (GABA reuptake) inhibitor | Focal epilepsy |
| Valproate | Multiple: ↑GAD activity, inhibits GABA-T, blocks Na⁺/Ca²⁺ channels | Epilepsy, bipolar disorder, migraine prophylaxis |
| Baclofen | GABA-B agonist | Muscle spasticity |
| Gabapentin/Pregabalin | Binds α2δ subunit of voltage-gated Ca²⁺ channels → reduces glutamate/substance P release (indirect GABA mimetic) | Neuropathic pain, epilepsy, anxiety |
The GABA–Glutamate Interplay
These two systems are deeply intertwined:
- GABA is synthesized from glutamate — the excitatory precursor directly generates the inhibitory neurotransmitter
- NMDA hypofunction on GABAergic interneurons (as in schizophrenia) causes disinhibition — paradoxically increasing downstream glutamate activity
- The E/I (excitation/inhibition) ratio is a central concept in epilepsy, schizophrenia, autism spectrum disorder, and anxiety
Sources: Stahl's Essential Psychopharmacology | Ganong's Review of Medical Physiology | Kaplan & Sadock's Comprehensive Textbook of Psychiatry | Neuroscience: Exploring the Brain | Goodman & Gilman's Pharmacological Basis of Therapeutics