Generate a short answer on this question for MBBS biochemistry exam based on Vasudevan with cycle
Asking for Preferences
Maple Syrup Urine Disease
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I have all the information needed for a complete, exam-ready answer. Here is the short answer on Maple Syrup Urine Disease for MBBS Biochemistry, based on Vasudevan-style with the metabolic cycle:
Maple Syrup Urine Disease (MSUD)
Definition
MSUD is a rare autosomal recessive inborn error of amino acid metabolism characterized by deficiency of the branched-chain α-keto acid dehydrogenase (BCKAD/BCKD) complex, resulting in accumulation of branched-chain amino acids (BCAAs) and their keto acids in blood and urine.
- Incidence: ~1:185,000 to 1:200,000 live births
Amino Acids Involved (BCAAs)
The three essential branched-chain amino acids affected are:
- Leucine (Leu)
- Isoleucine (Ile)
- Valine (Val)
Mnemonic: "LIV" (liver does NOT catabolize them - they are mainly catabolized in peripheral tissues/muscle)
Metabolic Pathway (The Cycle / Steps Blocked)
Normal BCAA catabolism proceeds in 3 steps:
BCAAs (Leucine, Isoleucine, Valine)
|
↓ Step 1: TRANSAMINATION
| (enzyme: Branched-chain aminotransferase; coenzyme: Pyridoxal phosphate / Vit B6)
|
α-Keto acids (α-ketoisocaproate, α-ketomethylvalerate, α-ketoisovalerate)
|
↓ Step 2: OXIDATIVE DECARBOXYLATION ← ❌ BLOCKED IN MSUD
| (enzyme: BCKD complex; coenzymes: TPP, Lipoic acid, FAD, NAD+, CoA)
|
Acyl CoA derivatives (Isovaleryl CoA, α-Methylbutyryl CoA, Isobutyryl CoA)
|
↓ Step 3: DEHYDROGENATION (analogous to β-oxidation of fatty acids)
|
End products:
Leucine → Acetoacetate + Acetyl CoA (KETOGENIC only)
Isoleucine → Acetyl CoA + Succinyl CoA (BOTH ketogenic + glucogenic)
Valine → Succinyl CoA (GLUCOGENIC only)
Key Enzyme Complex: BCKD
The BCKD complex has 4 subunits:
- E1α, E1β (branched-chain α-keto acid decarboxylase - uses TPP)
- E2 (dihydrolipoyl transacylase - uses Lipoic acid + CoA)
- E3 (dihydrolipoyl dehydrogenase - uses FAD + NAD+)
Important: The E3 subunit is shared with two other dehydrogenase complexes: Pyruvate dehydrogenase and α-Ketoglutarate dehydrogenase. Therefore, E3 deficiency causes a combined deficiency of all three complexes (lactic acidosis + MSUD features).
Pathophysiology
- Accumulated leucine is primarily neurotoxic - causes cerebral edema and neurological damage
- Accumulated α-keto acids are responsible for the characteristic maple syrup odor (especially due to isoleucine metabolite)
- Results in ketoacidosis (due to ketogenic nature of leucine and isoleucine)
Clinical Features
| Feature | Details |
|---|---|
| Onset | First few days of life (neonatal) |
| Odor | Maple syrup smell in urine, sweat, and earwax |
| Symptoms | Poor feeding, vomiting, lethargy, hypotonia |
| Neurological | Seizures, coma, decerebrate rigidity |
| Biochemistry | Ketonuria, metabolic acidosis (high anion gap) |
| If untreated | Fatal within weeks; survivors have severe intellectual disability |
Types of MSUD
- Classic form - Most severe; no BCKD activity; neonatal onset
- Intermediate form - Residual enzyme activity (up to 30%); milder symptoms
- Intermittent form - Episodes triggered by high protein intake or catabolic states
- Thiamine-responsive MSUD - Responds to pharmacological doses of Thiamine (Vit B1)
- E3-deficient form - Combined deficiency (MSUD + lactic acidosis)
Diagnosis
- Urine/plasma amino acid analysis: Marked elevation of leucine, isoleucine, valine + pathognomonic presence of L-alloisoleucine
- DNPH test (2,4-dinitrophenylhydrazine): Positive - characteristic precipitate (detects keto acids)
- Urine organic acids: Elevated branched-chain keto acids
- Newborn screening: Elevated leucine + isoleucine with normal phenylalanine
Treatment
- Diet: Synthetic formula free of all three BCAAs (Leu, Ile, Val), with controlled supplementation to allow normal growth
- Monitor leucine closely - it is the main cause of neurological damage
- Thiamine supplementation in responsive cases (pharmacological doses)
- Early diagnosis is essential - delayed treatment leads to irreversible intellectual disability
- Avoid catabolic states (illness, fasting) - catabolism of muscle protein releases BCAAs and worsens the disease
Exam Memory Points (Vasudevan Style)
| Point | Fact |
|---|---|
| Deficient enzyme | BCKD complex (branched-chain α-keto acid dehydrogenase) |
| Inheritance | Autosomal recessive |
| Amino acids | Leucine, Isoleucine, Valine (BCAAs) |
| Urine odor | Maple syrup (due to isoleucine metabolite) |
| Neurotoxic AA | Leucine |
| Shared subunit | E3 - shared with PDH and α-KGDH |
| Vitamin responsive variant | Thiamine (Vit B1) |
| DNPH test | Positive |
| Leucine end products | Acetoacetate + Acetyl CoA (pure ketogenic) |
| Valine end products | Succinyl CoA (pure glucogenic) |
| Isoleucine end products | Acetyl CoA + Succinyl CoA (both) |
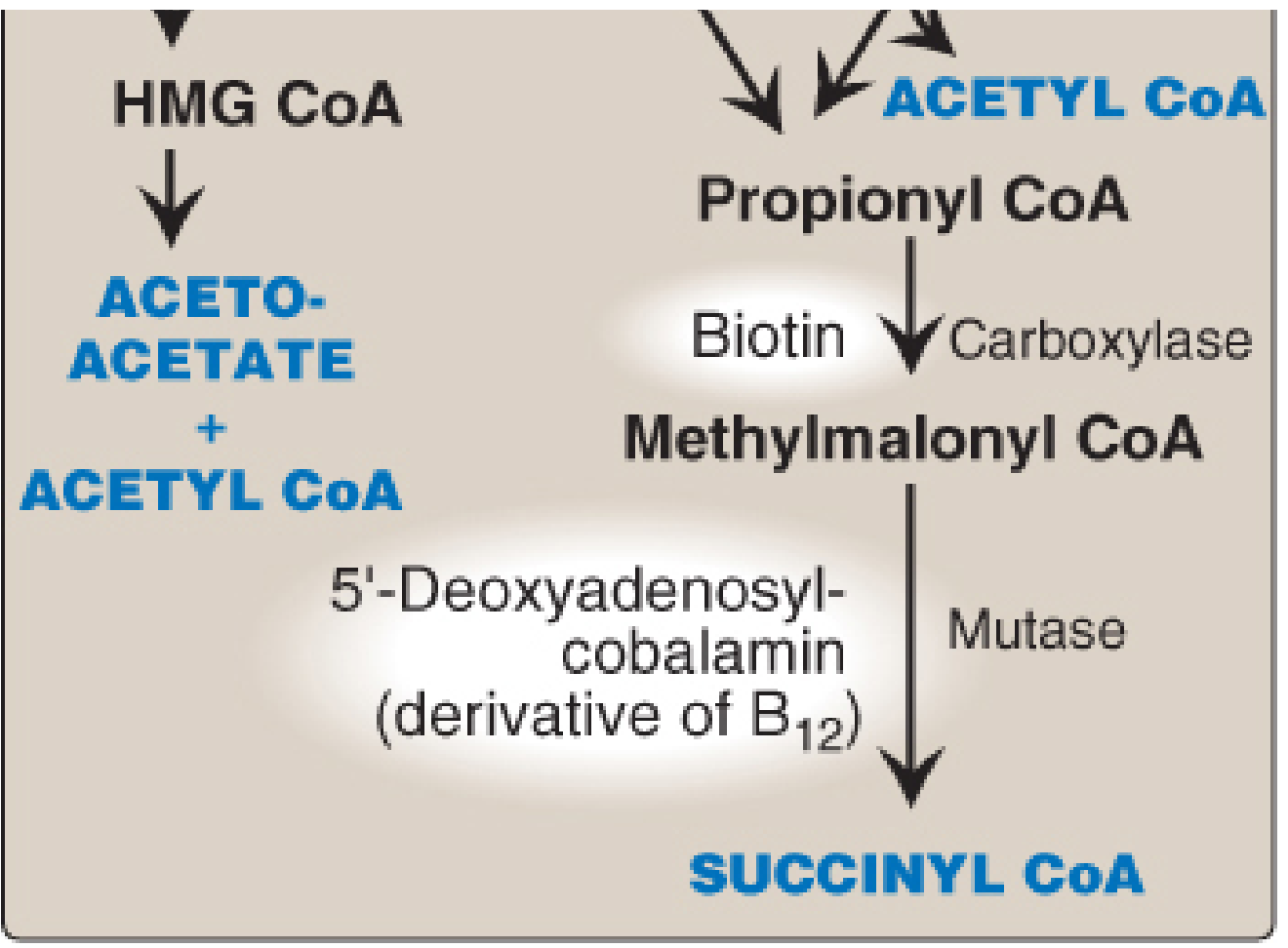
Figure: BCAA catabolism end products - Leucine → Acetoacetate + Acetyl CoA; Isoleucine → Acetyl CoA + Succinyl CoA; Valine → Succinyl CoA (via Propionyl CoA → Methylmalonyl CoA → Succinyl CoA). The BCKD step (oxidative decarboxylation) is blocked in MSUD. - Lippincott Illustrated Reviews Biochemistry, 8th ed.
Post-Transcriptional Modifications,One Carbon Metabolism, DNA Repair Mechanisms
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Here are the three complete short answers for your MBBS Biochemistry exam, based on Vasudevan/Lippincott style:
TOPIC 1: Post-Transcriptional Modifications of mRNA
Definition
Post-transcriptional modifications are processing events that convert the primary RNA transcript (pre-mRNA / hnRNA - heterogeneous nuclear RNA) into mature, functional mRNA before it exits the nucleus for translation.
These modifications occur in the nucleus and are specific to eukaryotes.
Overview Diagram
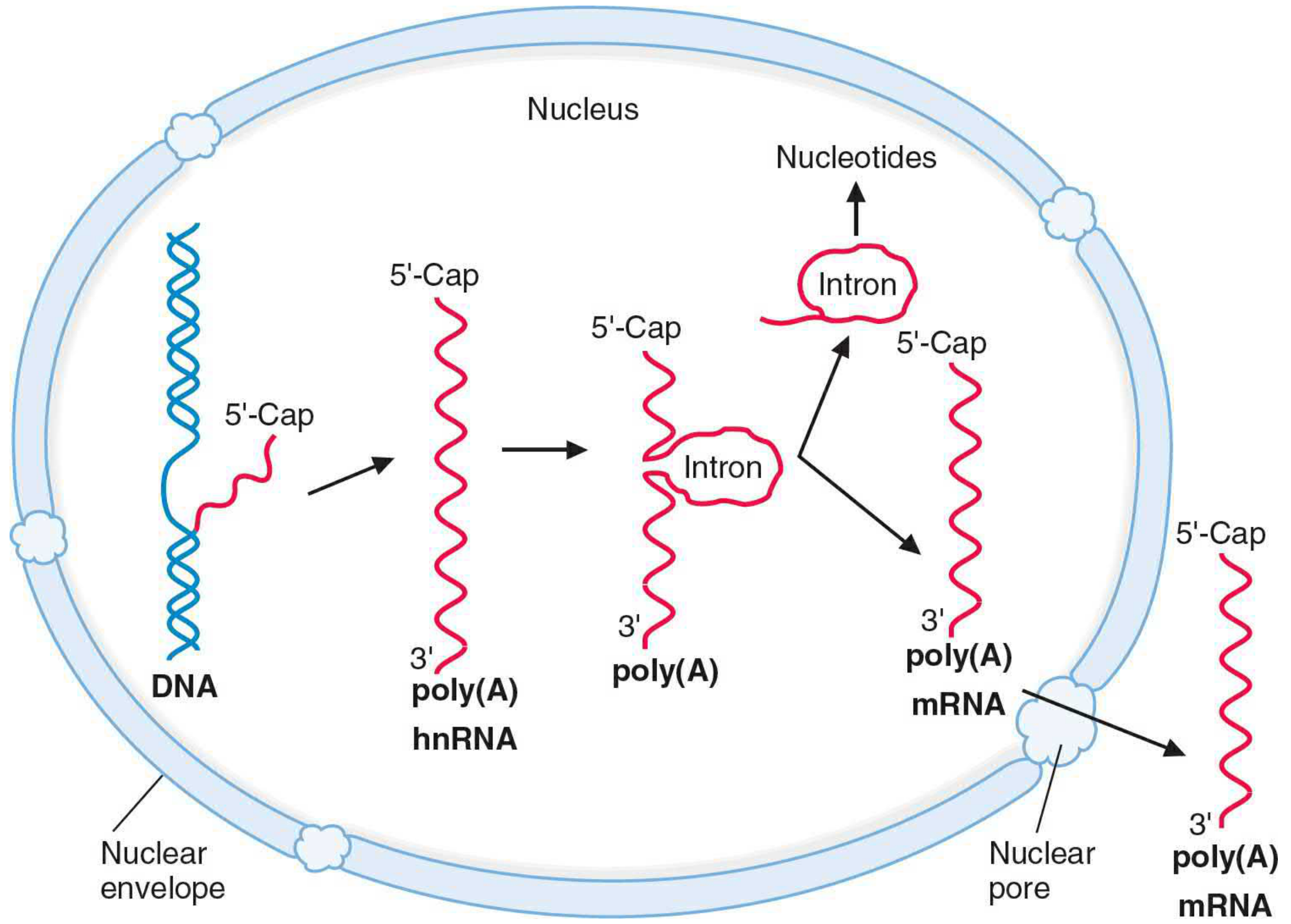
FIGURE: hnRNA processing - 5'-Cap addition, poly(A) tail addition, and splicing of introns to produce mature mRNA - Basic Medical Biochemistry, 6e
Three Major Modifications
1. Addition of 5' Cap (5'-Methyl Guanosine Cap)
- Added at the 5' end of hnRNA as it is being transcribed
- A guanosine triphosphate (GTP) is added to the 5' end via an unusual 5'-to-5' triphosphate linkage
- Methyl groups are donated by S-adenosylmethionine (SAM) to position N7 of guanine
- Three cap types:
- Cap 0: 7-methylguanosine only
- Cap 1: Cap 0 + methyl group on 2'-OH of N1 ribose
- Cap 2: Cap 1 + methyl group on 2'-OH of N2 ribose
- Functions:
- Protects mRNA from 5' exonuclease degradation
- Required for ribosome recognition/binding during translation initiation
- Facilitates nuclear export of mRNA
Clinical link: SAM is regenerated by reactions requiring folate and Vit B12. Hence folate/B12 deficiency impairs cap synthesis.
2. Addition of 3' Poly(A) Tail
- Added at the 3' end after transcription
- RNA polymerase reads past the stop codon until it encounters a polyadenylation signal: AAUAAA
- The primary transcript is cleaved 10-20 nucleotides downstream of this signal
- Poly(A) polymerase then adds ~200-250 adenine nucleotides to the 3' end (using ATP as substrate)
- No DNA template for the poly(A) tail - it is added post-transcriptionally
- Functions:
- Protects mRNA from 3' exonuclease degradation
- Helps export mRNA from nucleus
- Facilitates translation initiation
Clinical link: A mutation AAUAAA → AACAAA in β-globin gene disrupts the polyadenylation signal, producing only 1/10th normal β-globin mRNA, causing β-thalassemia.
3. Splicing - Removal of Introns (RNA Splicing)
- Pre-mRNA contains exons (expressed - present in mature mRNA) and introns (intervening sequences - removed)
- Consensus splice site sequences at intron boundaries are invariant:
- 5' splice site: GU (intron begins with GU)
- 3' splice site: AG (intron ends with AG)
- Rule: "GU-AG rule"
- Spliceosome (large ribonucleoprotein complex) mediates splicing
- Contains snRNPs (snurps) - small nuclear ribonucleoproteins rich in uracil: U1, U2, U4, U5, U6
- U1 binds near 5' exon/intron junction
- U2 binds within intron at an adenine-containing branch point
- Intron is removed as a lariat (loop) structure
| Component | Role |
|---|---|
| U1 snRNP | Binds 5' splice site |
| U2 snRNP | Binds branch point A in intron |
| U4/U5/U6 snRNP | Form spliceosome; catalyze splicing |
Alternative splicing: One gene can produce multiple proteins by including/excluding different exons (e.g., tropomyosin gene produces 5+ proteins).
Summary Table
| Modification | Location | Enzyme/Molecule | Function |
|---|---|---|---|
| 5' Cap | 5' end | Guanylyl transferase + SAM | Protects from degradation, ribosome binding |
| Poly(A) tail | 3' end | Poly(A) polymerase + ATP | Protects from degradation, stability |
| Splicing | Internal | Spliceosome (snRNPs) | Remove introns, join exons |
- Basic Medical Biochemistry - A Clinical Approach, 6e; Lippincott Illustrated Reviews Biochemistry, 8e
TOPIC 2: One Carbon Metabolism
Definition
One-carbon metabolism refers to reactions that transfer single carbon units (formyl, methylene, or methyl groups) from donors to acceptors. The primary carrier is Tetrahydrofolate (THF/FH4), derived from the vitamin folate.
The One-Carbon Pool Diagram
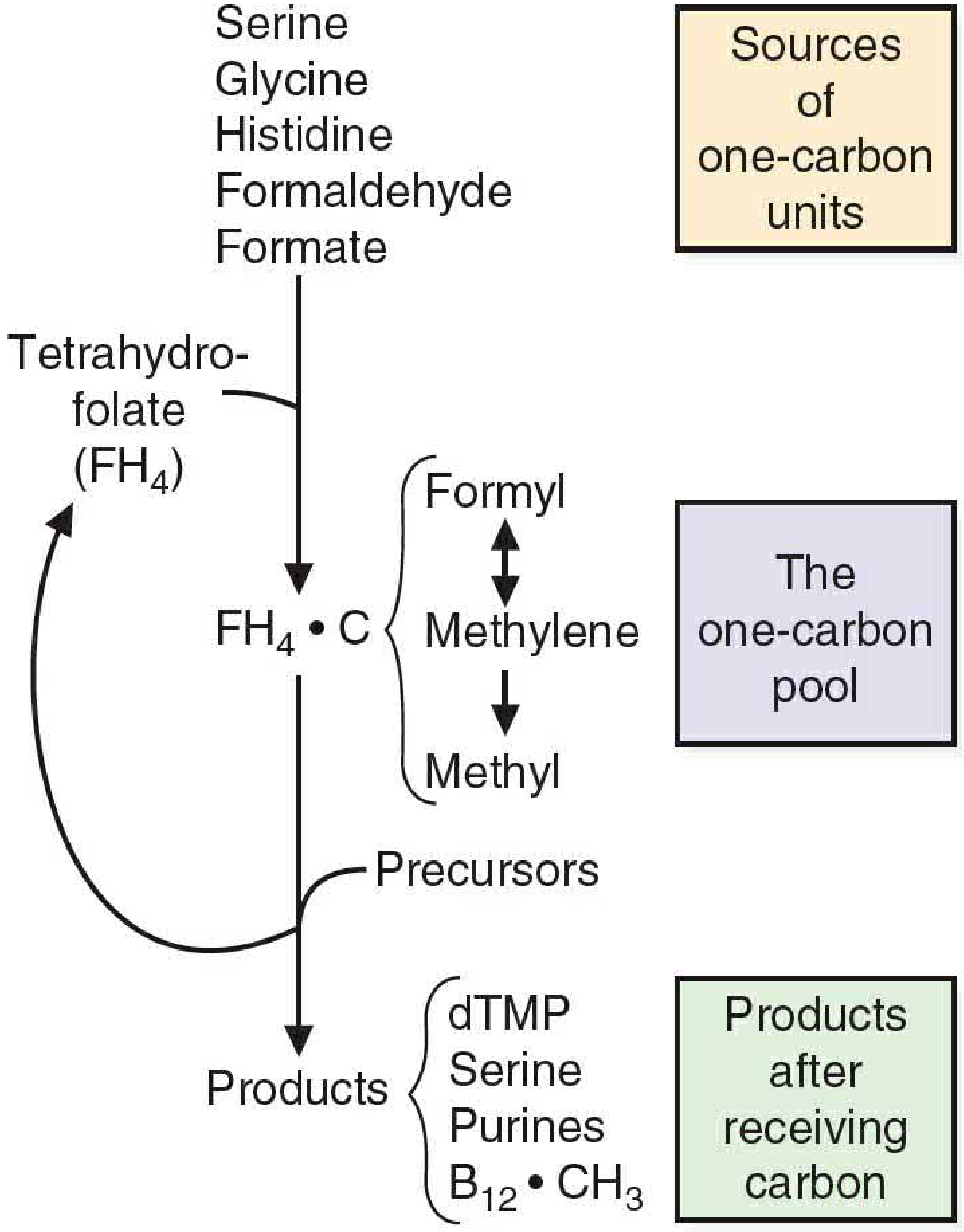
FIGURE 38.1: Overview of the one-carbon pool. Sources donate one-carbon units to FH4; products receive them. - Basic Medical Biochemistry, 6e
The Carrier: Tetrahydrofolate (THF / FH4)
- Derived from dietary folate (Vitamin B9) via dihydrofolate reductase
- Carbon is carried on N5 and/or N10 positions of the pteridine ring
- FH4 exists in three interconvertible forms carrying one-carbon units at different oxidation states:
| Form | Oxidation State | One-Carbon Group |
|---|---|---|
| N10-Formyl-FH4 | Most oxidized | -CHO (formyl) |
| N5,N10-Methylene-FH4 | Intermediate | -CH2- (methylene) |
| N5-Methyl-FH4 | Most reduced | -CH3 (methyl) |
- Conversion: Formyl ↔ Methylene → Methyl (one-way; methyl form is most stable = methyl trap)
Sources of One-Carbon Units (Donors to FH4)
| Amino Acid/Compound | Carbon Donated |
|---|---|
| Serine | Major donor; gives C3 to form glycine + N5,N10-methylene-FH4 |
| Glycine | Donates C2 via glycine cleavage system |
| Histidine | Donates C2 as formiminoglutamate (FIGLU) |
| Formaldehyde | Condenses with FH4 |
| Formate | Forms N10-formyl-FH4 |
FIGLU test: Histidine loading → FIGLU accumulates in urine in folate deficiency (Figlu excretion test used to diagnose folate deficiency)
Uses of One-Carbon Units (Products)
| Product | One-Carbon Group Used | FH4 Form |
|---|---|---|
| dTMP synthesis (from dUMP) | -CH2- (methylene) | N5,N10-Methylene-FH4 (oxidized to DHF in process) |
| Purine synthesis (C2 and C8 of purine ring) | -CHO (formyl) | N10-Formyl-FH4 |
| Serine synthesis (from glycine) | -CH2- (methylene) | N5,N10-Methylene-FH4 |
| Methionine regeneration (from homocysteine) | -CH3 (methyl) | N5-Methyl-FH4 + Vit B12 |
Role of Vitamin B12 and SAM
Methionine cycle / SAM cycle:
Homocysteine + N5-Methyl-FH4 → Methionine + FH4
(requires Vit B12 as cofactor; enzyme: Methionine synthase)
Methionine + ATP → SAM (S-Adenosylmethionine)
(universal methyl donor)
SAM donates methyl group to acceptors (creatine, phosphatidylcholine,
epinephrine, melatonin, methylated DNA, methylated histones)
SAM → SAH (S-Adenosylhomocysteine) → Homocysteine (cycle restarts)
Methyl Trap Hypothesis
- In Vitamin B12 deficiency: The enzyme methionine synthase (which uses B12) cannot function
- N5-Methyl-FH4 cannot donate its methyl group (this is the ONLY reaction that uses N5-methyl-FH4)
- Therefore, all FH4 gets "trapped" in the methyl form (most stable)
- Functional folate deficiency results despite normal total folate levels
- Cannot synthesize dTMP → cannot make DNA → Megaloblastic anemia
This explains why B12 deficiency causes megaloblastic anemia even though the direct cause is not lack of folate.
Clinical Significance
| Condition | Mechanism |
|---|---|
| Megaloblastic anemia | Folate or B12 deficiency → impaired dTMP/purine synthesis → impaired DNA synthesis in RBC precursors |
| Neural tube defects (spina bifida) | Folate deficiency in early pregnancy |
| Hyperhomocysteinemia | B12 or folate deficiency → homocysteine accumulates → cardiovascular and neurological disease |
| FIGLU-uria | Folate deficiency → histidine catabolism stalls → FIGLU excreted in urine |
| Methotrexate mechanism | Inhibits dihydrofolate reductase → blocks FH4 regeneration → blocks dTMP synthesis (anticancer) |
- Basic Medical Biochemistry - A Clinical Approach, 6e; Harper's Illustrated Biochemistry, 32e; Katzung Pharmacology 16e
TOPIC 3: DNA Repair Mechanisms
Definition
DNA repair refers to the cellular processes that identify and correct damage to DNA, which can result from imperfect DNA polymerase proofreading, normal chemical changes (spontaneous mutations), or environmental insults (radiation, mutagens).
DNA Repair Concept Map
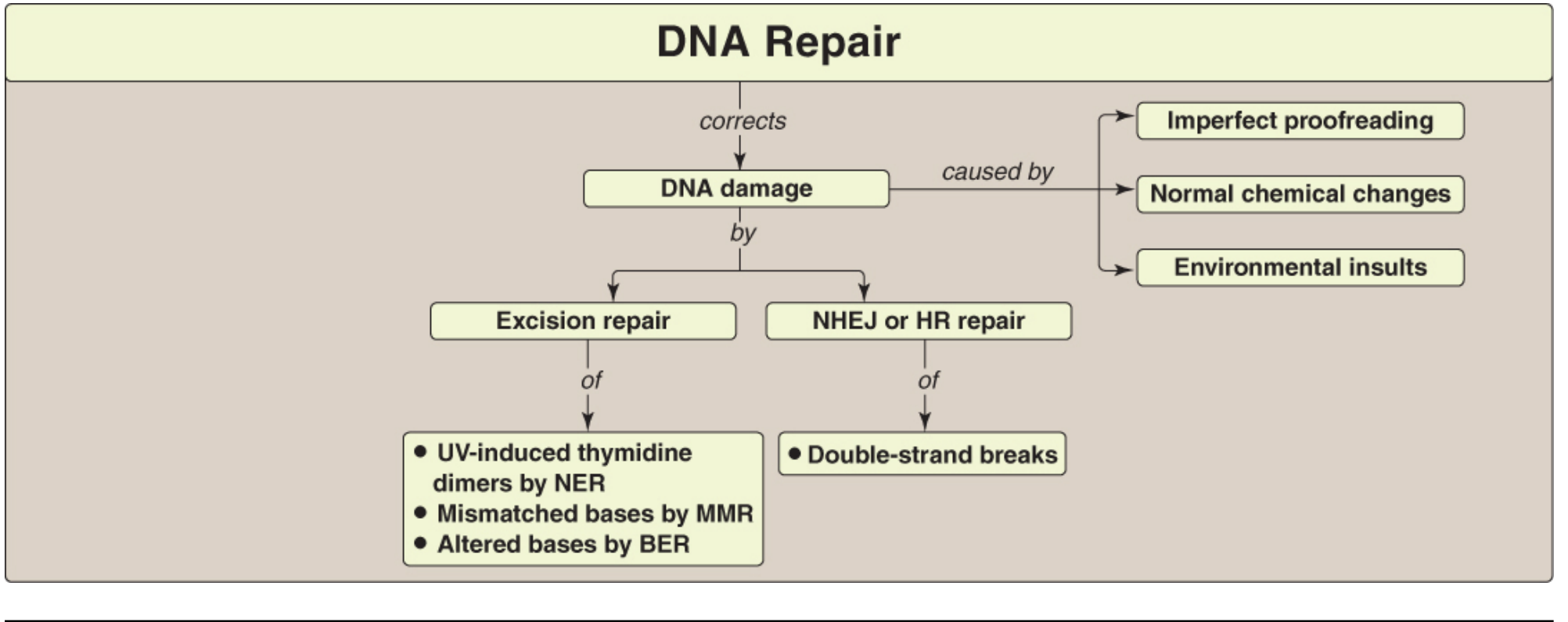
FIGURE 30.33: Key concept map for DNA repair - Lippincott Illustrated Reviews Biochemistry, 8e
Types of DNA Repair
1. Mismatch Repair (MMR)
| Feature | Detail |
|---|---|
| Damage repaired | Mismatched bases from DNA pol errors (e.g., G-T, A-C) |
| Mechanism | Mut proteins (MutS, MutL, MutH in bacteria) recognize mismatch; the newly synthesized (unmethylated) strand is excised and resynthesized correctly |
| Key proteins (human) | MSH2 and MLH1 (homologs of bacterial Mut proteins) |
| Clinical significance | Mutations in MSH2/MLH1 → Lynch syndrome (HNPCC - Hereditary Nonpolyposis Colorectal Cancer) |
| Timing | Occurs immediately after replication |
2. Nucleotide Excision Repair (NER)
| Feature | Detail |
|---|---|
| Damage repaired | Bulky lesions - pyrimidine dimers (T=T) caused by UV radiation; G adducts from benzo[a]pyrene (cigarette smoke) |
| Mechanism | A UV-specific endonuclease (uvrABC excinuclease) recognizes the bulky dimer → cleaves the damaged strand on both 5' and 3' sides → removes a short oligonucleotide containing the dimer → gap filled by DNA pol I + DNA ligase |
| Two subtypes | Global genomic NER (finds damage anywhere in genome); Transcription-coupled NER (repairs lesions blocking RNA polymerase) |
| Cell cycle | Occurs throughout the cell cycle |
| Clinical significance | Defects in XP proteins (XPA-XPG) required for NER → Xeroderma Pigmentosum (XP) - extreme UV sensitivity, multiple skin cancers |
XP: Autosomal recessive disorder. Inability to repair pyrimidine dimers. Patients develop freckles, severe photosensitivity, and early skin cancers.
3. Base Excision Repair (BER)
| Feature | Detail |
|---|---|
| Damage repaired | Altered single bases - deaminated bases (C→U, A→hypoxanthine), alkylated bases, lost bases (AP sites) |
| Step 1 | DNA glycosylase recognizes and removes the abnormal base by cleaving the N-glycosidic bond → creates an AP (apurinic/apyrimidinic) site |
| Step 2 | AP endonuclease cuts the phosphodiester backbone 5' to the AP site |
| Step 3 | deoxyribose phosphate lyase removes the base-free sugar phosphate |
| Step 4 | DNA pol I + DNA ligase fill the gap and seal |
| Common example | Cytosine spontaneously deaminates to Uracil (~10,000 times/cell/day) → BER removes U and inserts correct C |
Note: ~10,000 purine bases are lost per cell per day by spontaneous hydrolysis (apurinic sites) - repaired by BER.
4. Double-Strand Break (DSB) Repair
Two mechanisms repair double-strand breaks:
A. Non-Homologous End Joining (NHEJ)
- Proteins recognize, process, and directly ligate the broken DNA ends
- Some DNA is lost in the process
- Error-prone and mutagenic
- Occurs throughout the cell cycle (G1, S, G2)
- Defects: predisposition to cancer, immunodeficiency syndromes
- Cause of breaks: ionizing radiation, doxorubicin, free radicals
B. Homologous Recombination (HR)
- Uses enzymes from genetic recombination during meiosis
- Homologous DNA is used as template to replace lost sequences
- Error-free repair
- Occurs only in late S and G2 phases (when sister chromatid is available)
- Key proteins: BRCA1, BRCA2 (Breast Cancer 1, 2)
- Mutations in BRCA1/BRCA2 → increased risk of breast and ovarian cancer
Comparison Summary Table
| Repair Type | Damage Repaired | Key Enzyme/Protein | Clinical Disease |
|---|---|---|---|
| MMR | Mismatched bases (DNA pol errors) | MutS, MutL, MutH / MSH2, MLH1 | Lynch syndrome (HNPCC) |
| NER | Pyrimidine dimers, bulky adducts (UV, chemicals) | uvrABC excinuclease / XP proteins | Xeroderma Pigmentosum (XP) |
| BER | Altered/deaminated/lost bases, AP sites | DNA glycosylase, AP endonuclease, DNA pol I | - |
| NHEJ | Double-strand breaks | Ku proteins, DNA-PKcs | Immunodeficiency, cancer |
| HR | Double-strand breaks | BRCA1, BRCA2, RecA homologs | Breast/Ovarian cancer |
Exam Memory Mnemonics
-
"NER = UV light damage" - Nucleotide Excision Repair removes bulky UV dimers
-
"BER = Bad Base Removal" - Base Excision Repair removes chemically altered individual bases
-
"MMR = Mistakes Made during Replication" - corrects DNA pol errors
-
BRCA = NHEJ is error-prone, HR is error-FREE using homologous template
-
XP (Xeroderma Pigmentosum) = NER defect = can't fix UV damage = skin cancers
-
HNPCC/Lynch syndrome = MMR defect = colon cancer
-
Lippincott Illustrated Reviews Biochemistry, 8e; Basic Medical Biochemistry - A Clinical Approach, 6e; Thompson & Thompson Genetics, 9e
Respiratory Acidosis, Metabolic Acidosis
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
I now have all the information needed for both topics. Here are the complete exam-ready answers:
TOPIC 1: Respiratory Acidosis
Definition
Respiratory acidosis is a primary acid-base disorder characterized by:
- ↑ PaCO2 (hypercapnia) > 45 mmHg (primary abnormality)
- ↓ pH < 7.35 (acidemia)
- Compensatory ↑ HCO3- (secondary change by kidneys)
It results from impaired CO2 elimination due to hypoventilation (decreased alveolar ventilation).
Henderson-Hasselbalch Basis
CO2 + H2O ⇌ H2CO3 ⇌ H+ + HCO3-
pH = pKa + log [HCO3-] / (0.03 × PaCO2)
In respiratory acidosis:
PaCO2 ↑ → H2CO3 ↑ → H+ ↑ → pH ↓
Acid-Base Classification Diagram
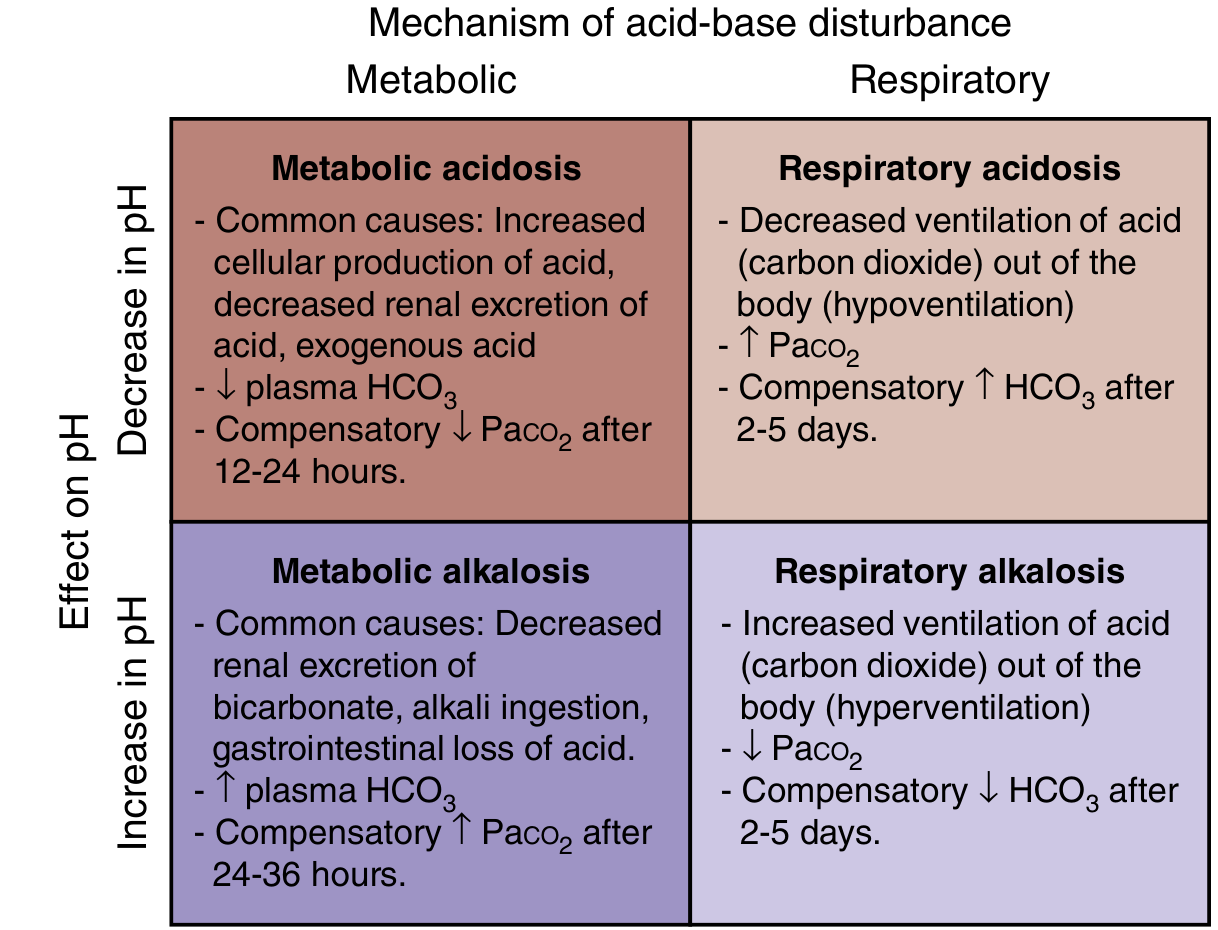
Fig. 113.1: Classification of the four major categories of acid-base disorders - Rosen's Emergency Medicine, 9e
Causes of Respiratory Acidosis
| Category | Examples |
|---|---|
| CNS depression (respiratory center) | Sedatives (opioids, benzodiazepines), barbiturates, general anesthesia, alcohol, head trauma, intracranial tumors |
| Neuromuscular disorders | Guillain-Barré syndrome, myasthenia gravis, poliomyelitis, motor neuron disease, hypokalemia |
| Chest wall disorders | Kyphoscoliosis, flail chest, obesity hypoventilation syndrome (Pickwickian syndrome) |
| Airway obstruction | Laryngospasm, bronchospasm, foreign body, sleep apnea |
| Lung parenchymal disease | Severe pneumonia, ARDS, pulmonary edema, severe COPD |
| Mechanical ventilation | Inadequate ventilator settings |
Mnemonic for causes: "CHAMPS" - CNS depression, Hypoventilation (muscles), Airway obstruction, Mechanical ventilation problems, Pulmonary disease, Sleep disorders
Compensation
| Type | Duration | Compensation Formula |
|---|---|---|
| Acute (cellular buffering) | Minutes to hours | ΔHCO3- = ΔPaCO2 × 0.1 (1 mEq/L rise per 10 mmHg ↑ PaCO2) |
| Chronic (renal) | >24-48 hours | ΔHCO3- = ΔPaCO2 × 0.4 (4 mEq/L rise per 10 mmHg ↑ PaCO2) |
- Renal compensation: kidneys ↑ H+ excretion (as NH4+ and titratable acid) + ↑ HCO3- reabsorption in proximal tubules
- Maximum HCO3- in compensation: ~38 mEq/L
- Respiratory compensation takes 2-5 days to fully develop
Laboratory Values
| Parameter | Acute Respiratory Acidosis | Chronic Respiratory Acidosis |
|---|---|---|
| pH | ↓ (< 7.35) | ↓ (less severe, near normal) |
| PaCO2 | ↑ (> 45 mmHg) | ↑ |
| HCO3- | Slight ↑ (~1 per 10 mmHg CO2) | Significant ↑ (~4 per 10 mmHg CO2) |
| Cl- | Normal | ↓ (excreted by kidneys to make room for HCO3-) |
Clinical Features
Due to hypercapnia (CO2 narcosis):
- Rapid rise in PaCO2: Anxiety, dyspnea, confusion, psychosis, hallucinations → coma
- Chronic hypercapnia: Sleep disturbances, memory loss, daytime somnolence, personality changes, tremor, myoclonus, asterixis
- Cerebral vasodilation by CO2: Headache, papilledema, focal weakness (mimics ↑ intracranial pressure)
- Respiratory signs: Cyanosis, accessory muscle use
Treatment
- Treat the underlying cause (primary goal)
- Improve ventilation: Bronchodilators (COPD), reversal agents (naloxone for opioids), non-invasive ventilation (BiPAP), mechanical ventilation if severe
- Bicarbonate only used cautiously in mixed metabolic + respiratory acidosis (target pH > 7.20, not normalization)
- Avoid rapid correction - sudden normalization of PaCO2 in chronic case can cause post-hypercapnic metabolic alkalosis
- Brenner & Rector's The Kidney; Sabiston Textbook of Surgery, 21e; Rosen's Emergency Medicine, 9e
TOPIC 2: Metabolic Acidosis
Definition
Metabolic acidosis is a primary acid-base disorder characterized by:
- ↓ HCO3- < 22 mEq/L (primary abnormality)
- ↓ pH < 7.35 (acidemia)
- Compensatory ↓ PaCO2 (respiratory compensation via hyperventilation)
Mechanisms (3 Ways HCO3- Falls)
- ↑ Endogenous acid production - exceeds kidney's ability to excrete (lactic acid, ketoacids)
- Loss of HCO3- - from GI tract (diarrhea) or kidneys (renal tubular acidosis)
- Inability of kidneys to excrete acid - progressive accumulation of endogenous acids (CKD, RTA)
Anion Gap (AG) - The Key Diagnostic Tool
Anion Gap = [Na+] - [Cl-] - [HCO3-]
Normal AG = 8-12 mEq/L (some sources: 3-11)
The AG represents unmeasured anions (mainly albumin, phosphate, sulfate).
Albumin correction: For every 1 g/dL drop in albumin below 4 g/dL, add 2.5 mEq/L to calculated AG.
Classification: High AG vs. Normal AG Metabolic Acidosis
A. High Anion Gap (HAGMA) - AG > 12 mEq/L
- Caused by accumulation of unmeasured organic/inorganic anions that consume HCO3-
- Chloride remains normal (normochloremic)
Mnemonic: "MUDPILES"
| Letter | Cause |
|---|---|
| M | Methanol |
| U | Uremia (renal failure) |
| D | Diabetic Ketoacidosis (DKA) |
| P | Paracetamol (acetaminophen), Paraldehyde, Propylene glycol |
| I | Isoniazid, Iron |
| L | Lactic acidosis (most common cause - ~50% of HAGMA) |
| E | Ethylene glycol, Ethanol |
| S | Salicylates |
B. Normal Anion Gap (NAGMA) / Hyperchloremic Acidosis - AG normal
- HCO3- is lost and replaced by Cl- (chloride rises proportionally)
- AG does not change
Mnemonic: "HARDUP"
| Letter | Cause |
|---|---|
| H | Hyperalimentation / Hospital-acquired (normal saline infusion) |
| A | Addison's disease (Acid infusion) / Acetazolamide (carbonic anhydrase inhibitors) |
| R | Renal Tubular Acidosis (Types 1, 2, 4) |
| D | Diarrhea (most common - HCO3- loss in stool) |
| U | Ureterosigmoidostomy |
| P | Pancreatic fistula/drainage |
Compensation (Respiratory Response)
Kussmaul breathing - deep, rapid breathing to blow off CO2 and raise pH
Winter's Formula (expected compensation):
Expected PaCO2 = 1.5 × [HCO3-] + 8 ± 2
OR ΔPaCO2 = 1.2 × ΔHCO3-
- Compensation begins within 12-24 hours
- If measured PaCO2 > expected → additional respiratory acidosis
- If measured PaCO2 < expected → additional respiratory alkalosis
Laboratory Values
| Parameter | Value |
|---|---|
| pH | ↓ < 7.35 |
| HCO3- | ↓ (primary - < 22 mEq/L) |
| PaCO2 | ↓ (compensatory hyperventilation) |
| Anion Gap | ↑ (HAGMA) or Normal (NAGMA) |
| Cl- | Normal (HAGMA) or ↑ (NAGMA = hyperchloremic) |
Clinical Features
| System | Feature |
|---|---|
| Respiratory | Kussmaul breathing (deep, sighing respirations - compensatory) |
| CVS | ↓ myocardial contractility, arrhythmias, blunted catecholamine response |
| CNS | Fatigue, confusion, lethargy, coma (severe) |
| Metabolic | Hyperkalemia (H+ shifts into cells, K+ shifts out) |
| Bones | Chronic acidosis causes bone demineralization (buffers H+) |
Kussmaul breathing is classically seen in DKA - fruity odor (acetone) + deep labored breathing + altered consciousness.
Delta Gap (Delta-Delta Ratio) - Detecting Mixed Disorders
Delta Gap = (Calculated AG - Normal AG) - (Normal HCO3- - Measured HCO3-)
Delta Gap > +6 → Concurrent Metabolic ALKALOSIS also present
Delta Gap < -6 → Concurrent Normal AG Metabolic ACIDOSIS also present
Treatment
| Approach | Detail |
|---|---|
| Treat underlying cause | Primary and most important |
| NaHCO3 (sodium bicarbonate) | Give when pH < 7.10 (severe) or pH < 7.20 with AKI; goal: raise pH above 7.20, not full normalization |
| Dialysis | In uremia, methanol/ethylene glycol poisoning |
| Insulin + fluids | DKA treatment |
| Lactic acidosis | Treat underlying cause (sepsis, shock) |
| Monitoring | Risk of hypokalemia when acidosis is corrected (K+ shifts back into cells) |
Quick Comparison Table: Respiratory vs. Metabolic Acidosis
| Feature | Respiratory Acidosis | Metabolic Acidosis |
|---|---|---|
| Primary defect | ↑ PaCO2 | ↓ HCO3- |
| pH | ↓ | ↓ |
| Cause | Hypoventilation | Acid gain / HCO3- loss / renal failure |
| Compensation | Renal ↑ HCO3- (2-5 days) | Respiratory ↓ PaCO2 (12-24 hrs) |
| Compensation formula | Acute: ΔHCO3- = 0.1×ΔPaCO2; Chronic: ΔHCO3- = 0.4×ΔPaCO2 | Expected PaCO2 = 1.5×HCO3- + 8 ± 2 |
| Breathing pattern | Slow/shallow (hypoventilation) | Deep/rapid (Kussmaul) |
| Key diagnosis tool | ABG: ↑ PaCO2 | Anion gap calculation |
| Key clinical sign | CO2 narcosis, papilledema | Kussmaul breathing, hyperkalemia |
- Brenner & Rector's The Kidney; Sabiston Textbook of Surgery 21e; Rosen's Emergency Medicine 9e
ELISA (Tumour/Immune functions), Xanthoma, Allopurinol, Translation — Drug Inhibition, Dietary Fibre, Cell Cycle Stage
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Loading Image
Here are all six complete MBBS Biochemistry exam answers:
TOPIC 1: ELISA (Enzyme-Linked Immunosorbent Assay)
Definition
ELISA (Enzyme-Linked Immunosorbent Assay) is a highly sensitive immunological technique that uses enzyme-labeled antibodies or antigens to detect and quantify specific antigens or antibodies in a sample. Results are measured by a visible color change produced when the enzyme acts on its substrate (read spectrophotometrically).
Principle
An antigen or antibody is immobilized onto a solid surface (microplate well). A complementary molecule tagged with an enzyme (e.g., Horseradish Peroxidase - HRP, or Alkaline Phosphatase - AP) is added. After washing, enzyme substrate is added. Enzyme converts the substrate → colored product detected by spectrophotometer.
ELISA Diagrams (Direct vs. Indirect)
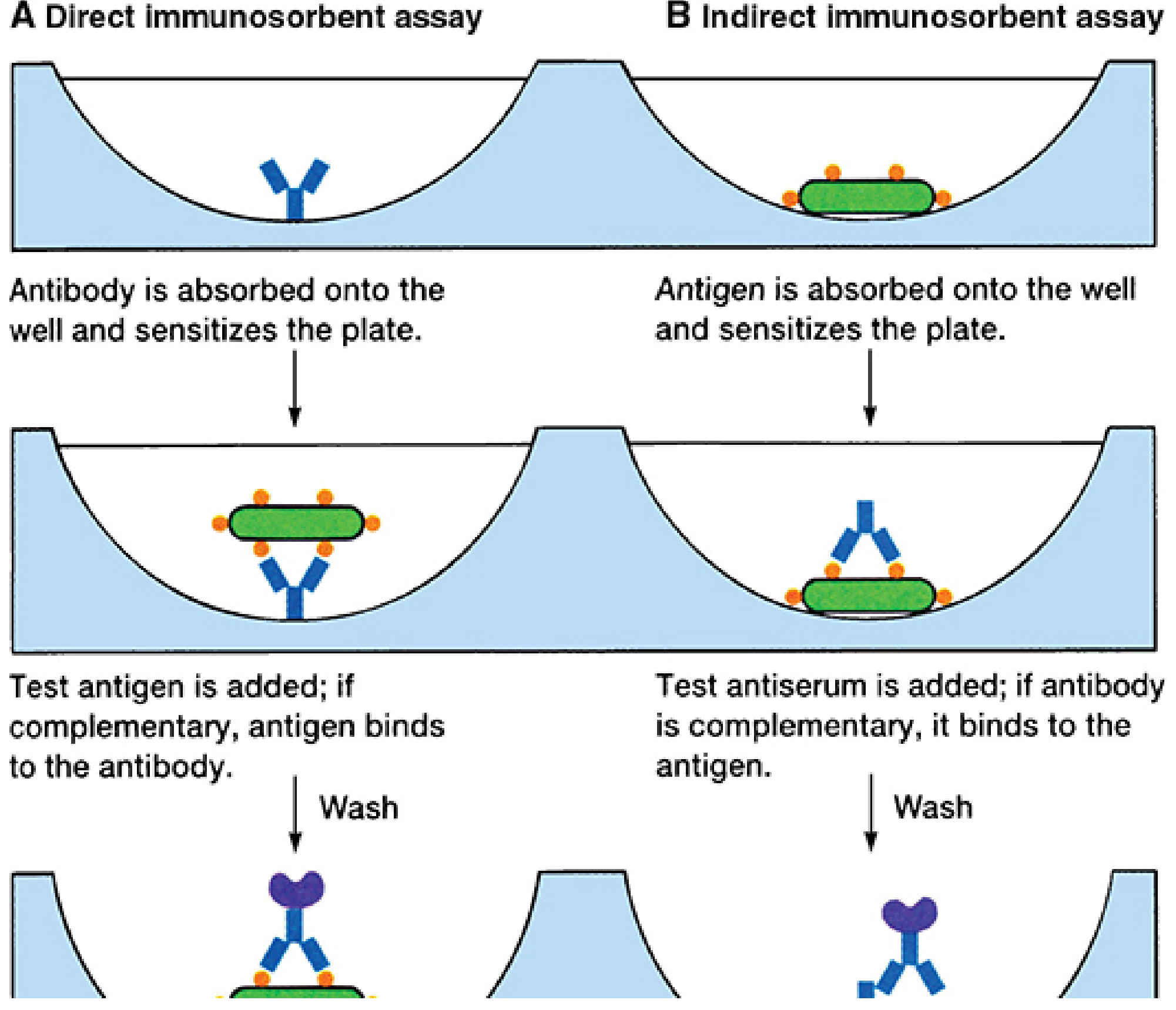
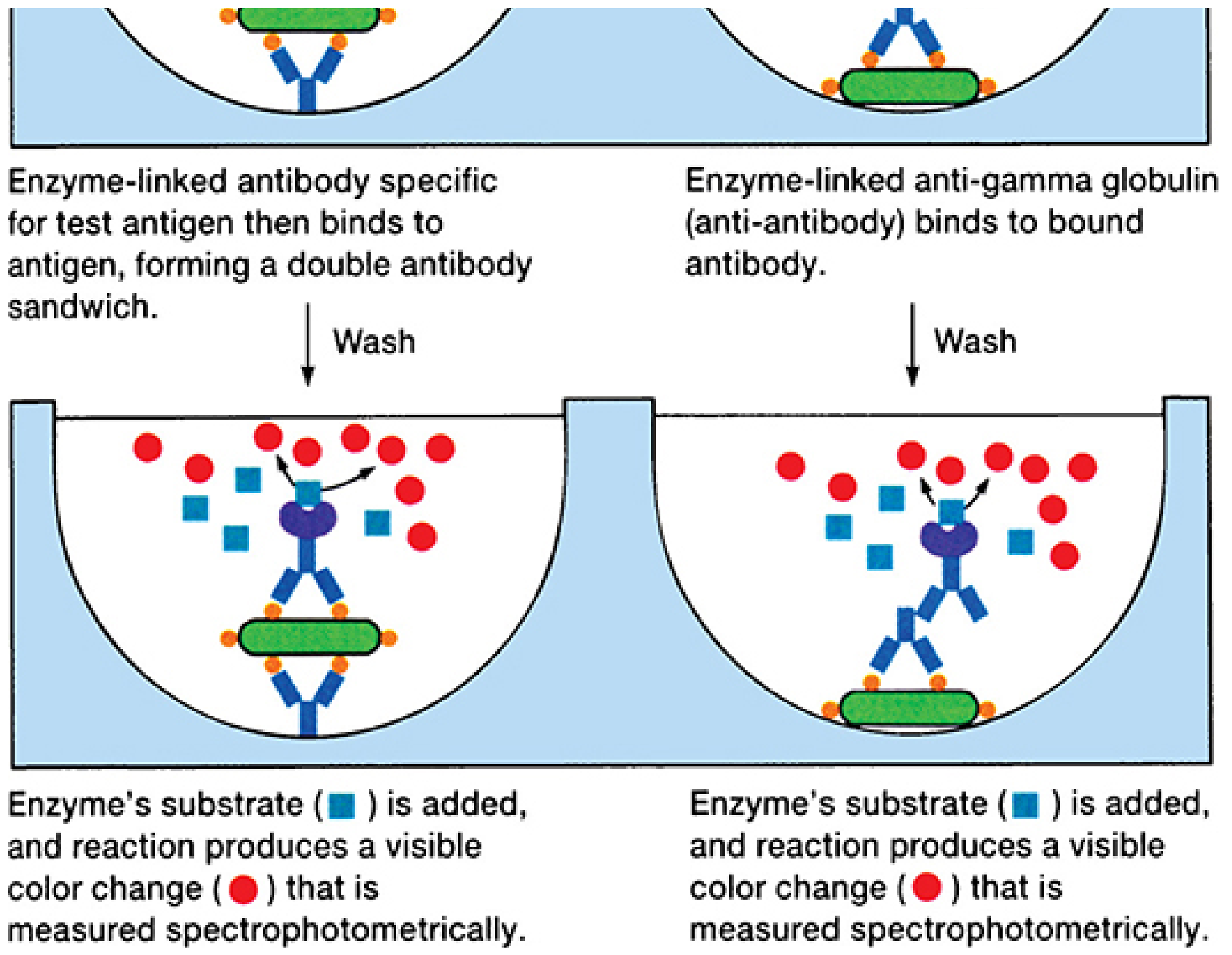
FIGURE 4-7: The ELISA/EIA test. A = Direct (double antibody sandwich) for antigen detection; B = Indirect for antibody detection. - Sherris & Ryan's Medical Microbiology, 8e
Types of ELISA
| Type | Principle | Detects | Use |
|---|---|---|---|
| Direct ELISA | Antigen coated on well; enzyme-labeled antibody added directly | Antigen | Antigen detection |
| Indirect ELISA | Antigen on well; test antibody added; enzyme-labeled anti-antibody (secondary Ab) added | Antibody | Antibody detection (e.g., HIV screening) |
| Sandwich ELISA | Capture antibody coated on well; antigen "sandwiched" between two antibodies; second Ab is enzyme-labeled | Antigen (between two Abs) | Tumor markers, hormones, cytokines |
| Competitive ELISA | Test antigen competes with enzyme-labeled antigen for fixed antibody | Low molecular weight antigens | Drug levels, haptens |
Enzymes Used
- Horseradish Peroxidase (HRP) - most common; substrate: TMB (3,3',5,5'-tetramethylbenzidine) → blue color → yellow when stopped
- Alkaline Phosphatase (AP) - substrate: p-nitrophenyl phosphate → yellow color
Applications
A. Tumor Markers (Cancer Diagnosis & Monitoring)
| Tumor Marker | Cancer | ELISA Used For |
|---|---|---|
| AFP (Alpha-fetoprotein) | Hepatocellular carcinoma, Germ cell tumors | Diagnosis, monitoring |
| PSA (Prostate-Specific Antigen) | Prostate cancer | Screening, monitoring |
| CEA (Carcinoembryonic Antigen) | Colorectal, lung, breast cancers | Monitoring recurrence |
| CA 125 | Ovarian cancer | Diagnosis, monitoring |
| CA 19-9 | Pancreatic cancer | Monitoring |
| β-hCG | Choriocarcinoma, testicular tumors | Diagnosis, monitoring |
| CA 15-3 | Breast cancer | Monitoring recurrence |
B. Immune Function / Infectious Disease
| Application | Example |
|---|---|
| HIV antibody screening | Indirect ELISA (initial test); confirmed by Western blot |
| Hepatitis B/C | HBsAg, anti-HCV antibody detection |
| Autoantibodies | Anti-dsDNA (SLE), Anti-CCP (RA), ANA, ANCA |
| Allergy testing | Specific IgE levels (allergens) |
| TORCH infections | IgM/IgG for Toxoplasma, Rubella, CMV, Herpes |
| Serum immunoglobulin levels | IgG, IgM, IgA quantification |
| Cytokines | IL-6, TNF-α levels in sepsis, inflammation |
Advantages
- High sensitivity and specificity
- Can process large batches (96-well plate format)
- Quantitative results (not just qualitative)
- No radioactive isotopes (unlike RIA)
- Relatively inexpensive and rapid
Limitations
-
Cannot detect very early infection (window period for HIV)
-
False positives possible (cross-reactions)
-
Requires known antigen/antibody for the assay to be designed
-
Indirect ELISA for HIV must be confirmed by Western blot
-
Sherris & Ryan's Medical Microbiology, 8e; Tietz Textbook of Laboratory Medicine, 7e
TOPIC 2: Xanthoma
Definition
Xanthomas are localized deposits of lipid-laden macrophages (foam cells) in the skin, tendons, and other tissues, resulting from hyperlipidemia. They appear as yellow plaques or nodules due to accumulated cholesterol esters and triglycerides.
Pathogenesis
Hyperlipidemia → elevated circulating lipoproteins → macrophages take up excess lipoproteins (especially LDL) via scavenger receptors → become foam cells → accumulate in skin/tendons → xanthoma
Types of Xanthoma
| Type | Location | Associated Lipid Disorder | Appearance |
|---|---|---|---|
| Xanthelasma | Periorbital (eyelids) | Familial hypercholesterolemia; may occur in normolipidemic individuals | Flat, yellow plaques near inner canthus |
| Tendinous xanthoma | Achilles tendon, extensor tendons of hands | Familial hypercholesterolemia (FH), dysbetalipoproteinemia | Firm, nodular swellings in tendons |
| Tuberous xanthoma | Elbows, knees, buttocks | Familial hypercholesterolemia, dysbetalipoproteinemia | Firm, yellow nodules over pressure points |
| Eruptive xanthoma | Buttocks, back, extensor surfaces | Hypertriglyceridemia (Type I, IV, V) | Sudden-onset yellow papules with red halo |
| Planar xanthoma | Palms, soles, trunk (plane xanthoma) | Dysbetalipoproteinemia (Type III), biliary obstruction | Yellow-orange flat plaques |
| Xanthoma striata palmaris | Creases of palms and fingers | Type III hyperlipidemia (pathognomonic) | Yellow streaks in palmar creases |
Lipid Disorder Association
| Xanthoma Type | Lipoprotein Elevated |
|---|---|
| Tendinous/Tuberous | LDL (Type IIa - Familial hypercholesterolemia) |
| Eruptive | Chylomicrons + VLDL (Type I, IV, V) |
| Planar/Xanthelasma | LDL or IDL (Type II, III) |
| Xanthoma striata palmaris | IDL (Type III - pathognomonic) |
Clinical Significance
- Xanthelasma and corneal arcus with tendinous xanthoma in a young person = strong indicator of Familial Hypercholesterolemia (FH)
- Homozygous FH: xanthomas in childhood; severe premature CAD
- Heterozygous FH: xanthomas by adulthood
- Eruptive xanthomas: appear suddenly with triglycerides >1000 mg/dL; risk of pancreatitis
- Xanthoma striata palmaris: pathognomonic for Type III hyperlipidemia (dysbetalipoproteinemia / broad-beta disease)
Treatment
-
Treat underlying hyperlipidemia (statins for LDL↑, fibrates/omega-3 for TG↑)
-
Surgical excision for cosmetic removal of xanthelasma (but recurs if lipid disorder untreated)
-
Dermatology 2-Volume Set, 5e; Henry's Clinical Diagnosis; Fitzpatrick's Dermatology
TOPIC 3: Allopurinol
Definition
Allopurinol is a xanthine oxidase inhibitor (purine analog) used as first-line urate-lowering therapy in the management of chronic gout and hyperuricemia.
Biochemical Background - Purine Catabolism
Adenine/Guanine (Purines)
↓
Hypoxanthine / Xanthine
↓ ← [Xanthine Oxidase] ← BLOCKED BY ALLOPURINOL
Uric Acid (insoluble - deposits in joints as monosodium urate crystals → GOUT)
Allopurinol competitively inhibits xanthine oxidase → blocks the last two steps in uric acid biosynthesis → hypoxanthine and xanthine accumulate (more soluble than uric acid) → excreted in urine without depositing in joints.
Mechanism of Action
- Allopurinol is a structural analog of hypoxanthine
- It is converted by xanthine oxidase to its active metabolite alloxanthine (oxypurinol)
- Both allopurinol and alloxanthine competitively inhibit xanthine oxidase
- Result: ↓ uric acid synthesis → ↓ serum urate → ↓ urate crystal deposition in joints, kidneys, and soft tissues
Pharmacokinetics
| Property | Detail |
|---|---|
| Route | Oral |
| Absorption | Completely absorbed |
| Active metabolite | Alloxanthine (Oxypurinol) - also a xanthine oxidase inhibitor |
| Half-life of metabolite | 15-18 hours |
| Dosing frequency | Once daily (due to long-acting metabolite) |
| Elimination | Renally excreted; dose reduction needed if eGFR < 30 mL/min/1.73m² |
Therapeutic Uses
- Chronic gout (prevention of attacks) - first-line, preferred over febuxostat and probenecid
- Hyperuricemia from malignancies (tumor lysis syndrome - after chemotherapy/radiation, large purine release)
- Uric acid nephrolithiasis (kidney stones)
- Lesch-Nyhan syndrome (HGPRT deficiency - severe hyperuricemia)
- Heart failure (urate lowering associated with better outcomes)
Important: Allopurinol is given for prevention, NOT for acute attacks. Starting allopurinol can actually precipitate an acute gout attack due to rapid changes in serum urate.
Adverse Effects
| Adverse Effect | Detail |
|---|---|
| Skin rash/hypersensitivity | Most common; risk ↑ in renal impairment; can progress to Stevens-Johnson syndrome (rare) |
| Acute gout flare | On initiation (use colchicine/NSAIDs prophylactically for 6 months) |
| GI upset | Nausea, diarrhea |
| Drug interaction | Inhibits metabolism of azathioprine and 6-mercaptopurine (both metabolized by xanthine oxidase) → dose reduction of these drugs required to avoid bone marrow suppression |
Comparison with Other Urate-Lowering Agents
| Drug | Mechanism | Notes |
|---|---|---|
| Allopurinol | XO inhibitor (purine analog) | First-line; dose adjust in renal disease |
| Febuxostat | XO inhibitor (non-purine) | Use when allopurinol is contraindicated; caution in cardiovascular disease |
| Probenecid | Uricosuric (blocks renal reabsorption of urate) | Avoid if creatinine clearance < 50 mL/min |
| Pegloticase | Recombinant uricase; converts urate to allantoin | For refractory gout |
| Colchicine | Tubulin depolymerization → ↓ neutrophil migration | Acute attack + prophylaxis |
- Lippincott Illustrated Reviews Pharmacology; Katzung Basic & Clinical Pharmacology, 16e
TOPIC 4: Translation - Drug Inhibition
Brief Review of Translation Steps
Translation occurs on ribosomes (70S in prokaryotes = 30S + 50S; 80S in eukaryotes = 40S + 60S) in three stages:
- Initiation: Ribosome assembles on mRNA at start codon (AUG); initiator tRNA (Met-tRNA) enters P site
- Elongation: Aminoacyl-tRNA enters A site → peptide bond formation (peptidyl transferase) → translocation (peptidyl-tRNA moves from A → P site, mRNA advances 3 nucleotides)
- Termination: Stop codon (UAA, UAG, UGA) reached; release factors → polypeptide released
Drugs That Inhibit Bacterial Translation (MBBS Key Table)
| Drug (Class) | Ribosomal Target | Mechanism | Effect |
|---|---|---|---|
| Streptomycin (Aminoglycoside) | 30S subunit (16S rRNA + 3 proteins) | Blocks initiation; causes mRNA misreading | Abnormal "streptomycin monosomes" form; premature termination; wrong amino acids incorporated |
| Tetracycline | 30S subunit | Blocks aminoacyl-tRNA binding to A site | Prevents elongation; reversible (bacteriostatic) |
| Chloramphenicol | 50S subunit | Blocks peptidyl transferase (inhibits peptide bond formation) | Prevents elongation; also inhibits mitochondrial protein synthesis → aplastic anemia |
| Erythromycin (Macrolide) | 50S subunit (23S rRNA) | Blocks translocation (A → P site movement) | Prevents elongation |
| Linezolid (Oxazolidinone) | 50S subunit | Blocks initiation (prevents formation of 70S initiation complex) | Prevents elongation start |
| Fusidic acid | EF-G (Elongation Factor G) | Prevents translocation | Blocks elongation |
Memory Tricks
"Buy AT 30, CELL at 50":
- 30S inhibitors: Buy = Bacteriostatic aminoglycosides, Aminoglycosides (streptomycin), Tetracyclines
- 50S inhibitors: Chloramphenicol, Erythromycin (macrolides), Linezolid, Lincosamides (clindamycin)
Key Clinical Notes
| Drug | Important Adverse Effect |
|---|---|
| Streptomycin | Ototoxicity (hearing loss) - limits use |
| Tetracycline | Resistance developing; chelates calcium (avoid in children < 8 yr, pregnancy - stains teeth, inhibits bone growth); photosensitivity |
| Chloramphenicol | Aplastic anemia (enters mitochondria); Gray baby syndrome (neonates - lack glucuronyl transferase); reserved for life-threatening infections only |
| Erythromycin | GI upset; QT prolongation; use for penicillin-allergic patients |
| Linezolid | Serotonin syndrome; thrombocytopenia; reserved for MRSA, VRE |
Eukaryotic Translation - Clinically Relevant Inhibitors
| Toxin/Drug | Target | Effect |
|---|---|---|
| Diphtheria toxin | EF-2 (Eukaryotic Elongation Factor 2) | ADP-ribosylates EF-2 → blocks translocation in eukaryotes |
| Ricin (castor bean) | 60S (28S rRNA) | Depurinates rRNA → irreversibly inactivates ribosome |
| Cycloheximide | 60S (80S) | Blocks translocation in eukaryotes (used in lab research) |
- Basic Medical Biochemistry - A Clinical Approach, 6e; Medical Microbiology, 9e; Katzung Pharmacology, 16e
TOPIC 5: Dietary Fibre
Definition
Dietary fiber is the edible part of plants that is nondigestible - comprising nonstarch polysaccharides and lignin (a noncarbohydrate polymer of aromatic alcohols). It resists digestion and absorption in the human small intestine.
Classification
| Type | Solubility | Examples | Action |
|---|---|---|---|
| Soluble fiber | Water-soluble; forms gel | Oat bran (beta-glucan), pectin (fruits), guar gum, psyllium, legumes | Fermented by gut bacteria in colon → Short-Chain Fatty Acids (SCFAs) |
| Insoluble fiber | Does not dissolve in water | Cellulose, hemicellulose, lignin (wheat bran, whole grains, vegetables) | Passes largely unchanged through GI tract |
Digestion and Metabolism
Dietary Fiber
↓ (resists small intestine digestion/absorption)
→ Colon → Gut microbiota → Fermentation
→ Short-Chain Fatty Acids (SCFAs): Acetate, Propionate, Butyrate
↓
SCFAs regulate:
- Host metabolism
- Immune system function
- Colonic cell proliferation (butyrate is preferred fuel for colonocytes)
Insoluble fiber: not fermented; absorbs 10-15× its weight in water → increases bowel motility
Health Benefits
| Benefit | Mechanism |
|---|---|
| ↓ Constipation | Insoluble fiber adds bulk, absorbs water, ↑ bowel motility (laxation) |
| ↓ LDL cholesterol | Soluble fiber binds bile acids in intestine → ↑ fecal bile acid excretion → liver uses more cholesterol to make bile → ↓ serum LDL (e.g., oat bran - 25-38 g/day reduces CHD risk) |
| ↓ Blood glucose spikes | Soluble fiber delays gastric emptying → blunts postprandial glucose rise (lowers Glycemic Index) |
| Satiety | Delayed gastric emptying → feeling full longer → helps weight management |
| ↓ Colon cancer risk | Dilutes carcinogens in stool; ↑ transit time; butyrate promotes colonocyte differentiation and apoptosis |
| ↓ Diverticulosis | Reduces intraluminal pressure by increasing stool bulk |
| ↓ Hemorrhoids | Easier defecation |
Recommended Intake (AI - Adequate Intake)
- Women: 25 g/day
- Men: 38 g/day
- Average American diet: only ~15 g/day (deficient)
Note: Fiber should be introduced gradually to the diet - sudden increase causes abdominal discomfort, gas, bloating, and diarrhea.
Glycemic Index and Fiber
- Glycemic Index (GI) ranks carbohydrate foods based on blood glucose response
- Fiber blunts the glycemic response by slowing gastric emptying and absorption
- Low GI (<55) foods are typically high in fiber
- Low-GI diets improve glycemic control in diabetics
Functional Fiber
Isolated fiber with proven health benefits (e.g., commercially available fiber supplements like psyllium husk). Has the same beneficial effects as dietary fiber.
Sources of Fiber
| Fiber Type | Food Sources |
|---|---|
| Soluble | Oats, barley, apples, oranges, legumes (lentils, beans), psyllium |
| Insoluble | Wheat bran, whole wheat bread, brown rice, vegetables (celery, carrots) |
- Lippincott Illustrated Reviews Biochemistry, 8e
TOPIC 6: Cell Cycle Stages
Definition
The cell cycle is the ordered sequence of events by which a cell duplicates its DNA and divides to produce two daughter cells. It coordinates DNA replication (S phase) and cell division (mitosis).
Cell Cycle Diagram
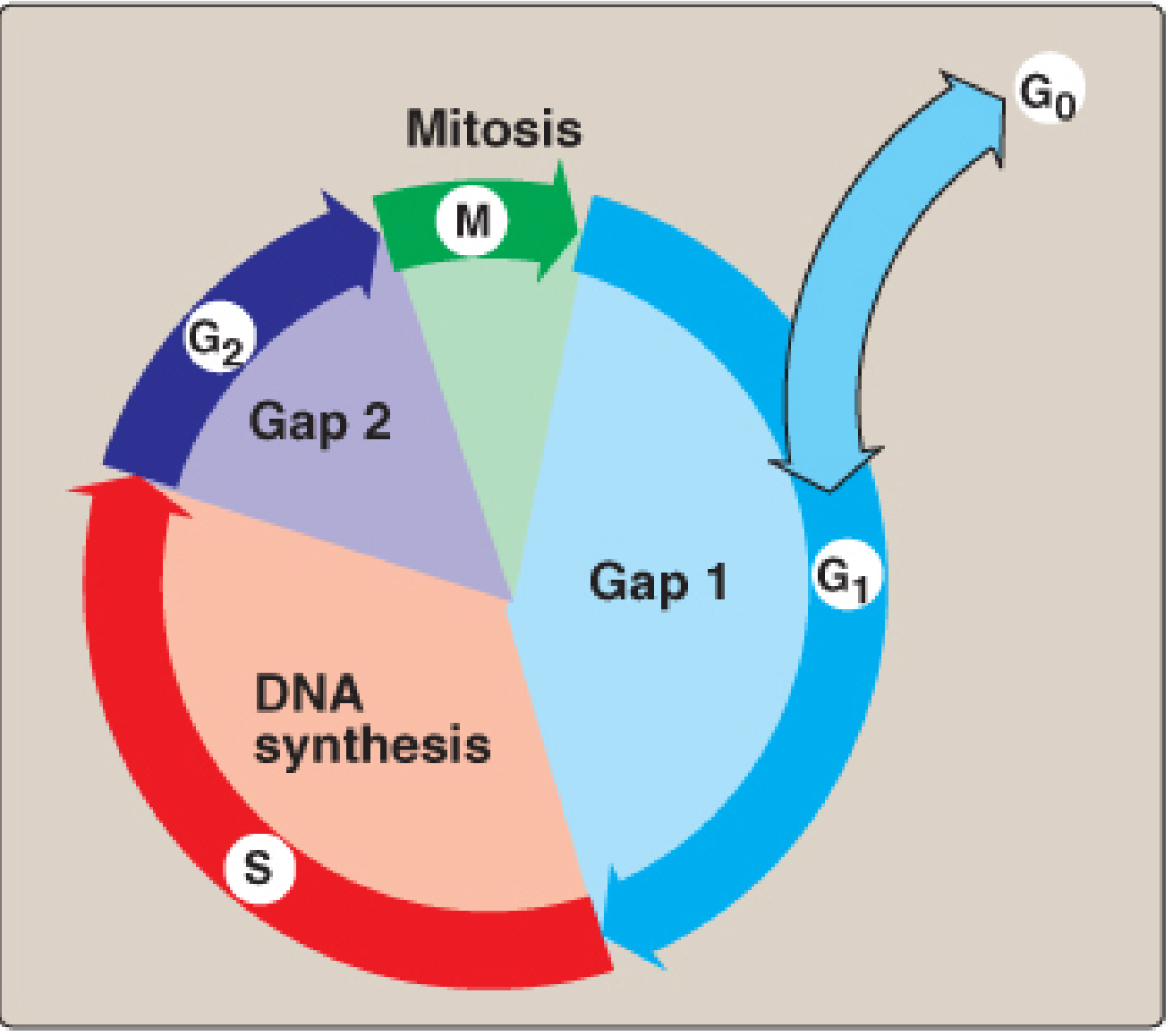
Figure 30.22: The eukaryotic cell cycle. Cells can leave the cycle and enter reversible quiescent state G0. - Lippincott Illustrated Reviews Biochemistry, 8e
Phases of the Cell Cycle
INTERPHASE (90% of total cycle time)
1. G1 Phase (Gap 1 / First Growth Phase)
- Period before DNA synthesis
- Cell grows in size; organelles duplicated
- Protein and RNA synthesis ↑
- Metabolically active: producing materials for DNA replication
- DNA content: 2N (diploid)
- Key checkpoint: G1/S checkpoint (Restriction point) - decides whether cell will divide
- Cells that stop dividing exit to G0 from G1
2. S Phase (Synthesis Phase)
- DNA replication occurs
- Each chromosome is replicated to form 2 sister chromatids
- DNA content doubles: 2N → 4N
- Histone synthesis also occurs (for packaging new DNA)
- Duration: ~6-8 hours
3. G2 Phase (Gap 2 / Second Growth Phase)
- Period after DNA synthesis, before mitosis
- Cell checks DNA was replicated correctly
- Proteins for mitosis (e.g., tubulin for spindle) are synthesized
- DNA content: 4N (tetraploid)
- Key checkpoint: G2/M checkpoint - ensures DNA is intact before division
M PHASE (Mitosis) - ~1 hour
Cell division occurs in 4 sequential sub-stages:
| Sub-stage | Key Events |
|---|---|
| Prophase | Chromosomes condense; mitotic spindle forms; nuclear envelope breaks down |
| Metaphase | Chromosomes align at metaphase plate (equatorial plane); spindle fibers attach to kinetochores |
| Anaphase | Sister chromatids separate and move to opposite poles |
| Telophase | Nuclear envelope reforms; chromosomes decondense; two nuclei form |
| Cytokinesis | Cytoplasm divides; two daughter cells formed (each 2N) |
G0 Phase (Quiescence)
- Cells that have exited the cell cycle
- Non-dividing, resting state (reversible)
- Examples: mature T lymphocytes, neurons, hepatocytes (can be stimulated back)
- Permanently non-dividing cells (terminally differentiated): neurons, cardiac muscle cells, skeletal muscle cells
Cell Cycle Checkpoints
| Checkpoint | Location | Function | Key Molecules |
|---|---|---|---|
| G1/S checkpoint (Restriction point) | Late G1 | Checks: cell size, nutrients, growth factors, DNA integrity | Cyclin D + CDK4/6; Rb protein; p53 |
| G2/M checkpoint | G2 | Checks: DNA fully replicated, DNA damage repaired | Cyclin B + CDK1 (MPF - Maturation Promoting Factor) |
| Spindle assembly checkpoint | Metaphase | Checks: all chromosomes attached to spindle | Securin, Separase, APC/C |
Regulators of the Cell Cycle
| Molecule | Role |
|---|---|
| Cyclins | Regulatory proteins; levels rise and fall through cycle; activate CDKs |
| CDKs (Cyclin-Dependent Kinases) | Catalytic kinase subunit; active only when bound to cyclin |
| Cyclin D + CDK4/6 | Drive through G1; phosphorylate Rb |
| Cyclin E + CDK2 | G1/S transition |
| Cyclin A + CDK2 | S phase progression |
| Cyclin B + CDK1 | G2/M transition (MPF) |
| p53 | Tumor suppressor; activates p21 (CDK inhibitor); stops cell cycle at G1 checkpoint after DNA damage |
| Rb (Retinoblastoma protein) | Tumor suppressor; inhibits G1 progression; phosphorylated by Cyclin D/CDK4 to release the brake |
DNA Content Through the Cell Cycle
G1: 2N (diploid)
S: 2N → 4N (synthesis in progress)
G2: 4N (tetraploid)
M: 4N → 2N + 2N (each daughter cell diploid)
Clinical Relevance: Cell Cycle and Cancer Drugs
| Drug | Cell Cycle Phase Targeted | Mechanism |
|---|---|---|
| Methotrexate, 5-FU | S phase | Inhibit DNA/thymidylate synthesis |
| Hydroxyurea | S phase | Inhibits ribonucleotide reductase |
| Bleomycin | G2/M | Causes DNA strand breaks |
| Vinca alkaloids (vincristine) | M phase | Inhibit tubulin polymerization → block spindle |
| Taxanes (paclitaxel) | M phase | Stabilize tubulin → prevent spindle disassembly |
| Colchicine | M phase | Depolymerizes tubulin → blocks spindle formation |
- Lippincott Illustrated Reviews Biochemistry, 8e; Basic Medical Biochemistry - A Clinical Approach, 6e; Quick Compendium of Clinical Pathology, 5e
give summary for all these questions
Here is a Quick Revision Summary of all topics covered in this session:
MBBS Biochemistry - Master Summary Sheet
1. Maple Syrup Urine Disease (MSUD)
- Defect: BCKD complex (Branched-chain α-keto acid dehydrogenase)
- Inheritance: Autosomal recessive
- Affected AAs: Leucine, Isoleucine, Valine (BCAAs)
- Urine odor: Maple syrup (due to isoleucine metabolite)
- Neurotoxic: Leucine (cerebral edema)
- BCKD cofactors: TPP, Lipoic acid, FAD, NAD+, CoA
- E3 subunit shared with PDH and α-KGDH complexes
- Leucine end products: Acetoacetate + Acetyl CoA (purely ketogenic)
- Valine: Succinyl CoA (glucogenic); Isoleucine: Both
- Dx: Plasma AA analysis, DNPH test positive, L-alloisoleucine pathognomonic
- Tx: BCAA-free synthetic diet; thiamine in responsive variants
2. Post-Transcriptional Modifications (mRNA Processing)
Three modifications in nucleus convert hnRNA → mature mRNA:
| Modification | End | Enzyme/Donor | Function |
|---|---|---|---|
| 5' Cap (7-methylguanosine) | 5' end | Guanylyl transferase + SAM | Protects, ribosome recognition |
| Poly(A) tail (~200-250 A's) | 3' end | Poly(A) polymerase + ATP | Protects, stability |
| Splicing (intron removal) | Internal | Spliceosome (U1,U2,U4,U5,U6 snRNPs) | Joins exons, GU-AG rule |
- β-thalassemia clinical link: AAUAAA → AACAAA mutation destroys polyadenylation signal
- SAM for cap synthesis requires Folate + Vit B12 for regeneration
3. One Carbon Metabolism
- Carrier: Tetrahydrofolate (FH4) derived from Folate (Vit B9)
- Carbon carried on: N5 and/or N10 positions
- Three forms: N10-Formyl (most oxidized) ↔ N5,N10-Methylene ↔ N5-Methyl (most reduced/stable)
- Sources: Serine (major), Glycine, Histidine, Formate, Formaldehyde
- Products: dTMP (methylene-FH4), Purine C2 & C8 (formyl-FH4), Methionine (methyl-FH4 + B12)
- SAM (S-adenosylmethionine): universal methyl donor → creatine, phosphatidylcholine, epinephrine, DNA methylation
- Methyl Trap: B12 deficiency → N5-methyl-FH4 accumulates → functional folate deficiency → megaloblastic anemia
- FIGLU test: Histidine loading → FIGLU in urine = folate deficiency
4. DNA Repair Mechanisms
| Type | Damage | Key Proteins | Clinical Disease |
|---|---|---|---|
| MMR (Mismatch Repair) | DNA pol errors (mismatched bases) | MSH2, MLH1 | Lynch syndrome / HNPCC |
| NER (Nucleotide Excision Repair) | UV-induced pyrimidine dimers; bulky adducts | XP proteins (XPA-XPG), uvrABC | Xeroderma Pigmentosum |
| BER (Base Excision Repair) | Deaminated/alkylated bases, AP sites | DNA glycosylase, AP endonuclease | - |
| NHEJ | Double-strand breaks | Ku proteins | Cancer, immunodeficiency |
| HR | Double-strand breaks | BRCA1, BRCA2 | Breast/Ovarian cancer |
- BER steps: Glycosylase removes base → AP endonuclease cuts → lyase removes sugar → DNA pol I fills gap → ligase seals
- NHEJ = error prone; HR = error free (uses homologous template, occurs in S/G2)
5. Respiratory Acidosis
| Feature | Value |
|---|---|
| Primary defect | ↑ PaCO2 > 45 mmHg (hypoventilation) |
| pH | ↓ < 7.35 |
| Compensation | Renal ↑ HCO3- (takes 2-5 days) |
| Acute formula | ΔHCO3- = ΔPaCO2 × 0.1 |
| Chronic formula | ΔHCO3- = ΔPaCO2 × 0.4 |
| Key symptoms | CO2 narcosis: confusion, asterixis, headache, papilledema, coma |
| Causes | COPD, opioids, neuromuscular disease, obesity-hypoventilation |
| Treatment | Treat cause; improve ventilation; NaHCO3 only in mixed acidosis |
6. Metabolic Acidosis
| Feature | Value |
|---|---|
| Primary defect | ↓ HCO3- < 22 mEq/L |
| pH | ↓ < 7.35 |
| Compensation | Respiratory ↓ PaCO2 (12-24 hrs) - Kussmaul breathing |
| Winter's formula | Expected PaCO2 = 1.5 × HCO3- + 8 ± 2 |
| AG formula | Na+ - Cl- - HCO3- (normal = 8-12) |
| High AG causes | MUDPILES (Methanol, Uremia, DKA, Paracetamol, Isoniazid, Lactic acidosis, Ethylene glycol, Salicylates) |
| Normal AG causes | HARDUP (Hyperalimentation, Addison's/Acetazolamide, RTA, Diarrhea, Ureterosigmoidostomy, Pancreatic fistula) |
| Key sign | Kussmaul breathing; hyperkalemia |
| Treatment | Treat cause; NaHCO3 if pH < 7.10 |
7. ELISA
| Type | Plate coated with | Detects | Use |
|---|---|---|---|
| Direct (Sandwich) | Capture antibody | Antigen | Tumor markers (AFP, PSA, CEA, CA125) |
| Indirect | Antigen | Antibody | HIV screening, autoantibodies |
| Competitive | Antibody | Antigen (by competition) | Drug levels, haptens |
- Enzymes: HRP (TMB substrate → blue), Alkaline Phosphatase (p-nitrophenyl phosphate → yellow)
- HIV ELISA: indirect; must confirm with Western blot
- Tumor markers: AFP (HCC), PSA (prostate), CEA (colorectal), CA-125 (ovarian), β-hCG (choriocarcinoma)
8. Xanthoma
| Type | Location | Lipid Disorder |
|---|---|---|
| Xanthelasma | Eyelids | FH; may be normolipidemic |
| Tendinous | Achilles, extensor tendons | Familial hypercholesterolemia (↑ LDL) |
| Eruptive | Buttocks, back | Hypertriglyceridemia (↑ TG) |
| Tuberous | Elbows, knees | FH / dysbetalipoproteinemia |
| Xanthoma striata palmaris | Palm creases | Pathognomonic for Type III (dysbetalipoproteinemia) |
- Pathogenesis: Hyperlipidemia → macrophage foam cells in skin/tendons
- Treatment: Statin (↑LDL), Fibrate (↑TG)
9. Allopurinol
- Class: Xanthine oxidase inhibitor (purine analog, structural analog of hypoxanthine)
- Mechanism: Blocks last 2 steps of uric acid synthesis; converted to active metabolite alloxanthine (oxypurinol)
- Used for: Chronic gout prevention, tumor lysis syndrome, uric acid kidney stones
- Dosing: Once daily (alloxanthine t½ = 15-18 hrs); renally cleared - dose reduce if eGFR <30
- Key ADR: Skin rash/hypersensitivity (most common); starts gout flare (add colchicine for 6 months)
- Critical drug interaction: Inhibits xanthine oxidase → cannot metabolize azathioprine / 6-MP → dose reduce by 75% to avoid bone marrow suppression
- Compare: Febuxostat (non-purine XO inhibitor); Probenecid (uricosuric); Colchicine (anti-inflammatory, not urate-lowering)
10. Translation - Drug Inhibition
"Buy AT 30, CELL at 50":
| Drug | Target | Mechanism | ADR |
|---|---|---|---|
| Streptomycin | 30S | Blocks initiation; mRNA misreading | Ototoxicity |
| Tetracycline | 30S | Blocks aminoacyl-tRNA to A site (bacteriostatic, reversible) | Teeth staining, avoid in children <8yr |
| Chloramphenicol | 50S | Blocks peptidyl transferase (peptide bond) | Aplastic anemia, Gray baby syndrome |
| Erythromycin | 50S (23S rRNA) | Blocks translocation (A→P site) | GI upset; use in penicillin allergy |
| Linezolid | 50S | Blocks initiation (70S complex) | Serotonin syndrome, thrombocytopenia |
| Diphtheria toxin | EF-2 (eukaryotic) | ADP-ribosylates EF-2; blocks translocation | Diphtheria myocarditis, neuropathy |
11. Dietary Fibre
| Property | Soluble Fiber | Insoluble Fiber |
|---|---|---|
| Examples | Oat bran, pectin, guar gum, legumes | Cellulose, wheat bran, lignin |
| In colon | Fermented → SCFAs (acetate, propionate, butyrate) | Passes unchanged |
| Key benefit | ↓ LDL (↑ bile acid excretion), ↓ blood glucose spikes, satiety | ↓ Constipation, ↓ diverticulosis, ↓ colon cancer |
- Recommended intake: 25 g/day (women), 38 g/day (men); average ~15 g/day
- Absorbs 10-15× its weight in water → bulk → laxation
- Butyrate = preferred fuel for colonocytes; promotes apoptosis of colon cancer cells
- Introduce gradually - sudden increase → gas, bloating
12. Cell Cycle Stages
G0 ← Exit ←─── G1 ──→ S ──→ G2 ──→ M ──→ (repeat)
(quiescence) (2N) (2N→4N) (4N) (4N→2N+2N)
| Phase | DNA | Key Event | Duration |
|---|---|---|---|
| G1 | 2N | Cell growth, protein/RNA synthesis | Longest (variable) |
| S | 2N→4N | DNA replication, histone synthesis | ~6-8 hours |
| G2 | 4N | Spindle protein synthesis, DNA check | ~4-5 hours |
| M (Mitosis) | 4N→2N | Prophase→Metaphase→Anaphase→Telophase→Cytokinesis | ~1 hour |
| G0 | 2N | Quiescent; neurons, mature RBCs, cardiac muscle | Indefinite |
Checkpoints:
- G1/S (Restriction point): Cyclin D + CDK4/6; Rb; p53 → mutated in most cancers
- G2/M: Cyclin B + CDK1 (MPF)
- Spindle: APC/C complex
Cancer drugs by phase: Methotrexate/5-FU (S phase) → Bleomycin (G2) → Vinca alkaloids/Taxanes/Colchicine (M phase)
Exam tip: These 12 topics cover amino acid disorders, molecular biology (transcription/translation/DNA repair), acid-base physiology, lipid disorders, pharmacology, and nutrition - all high-yield areas for MBBS Biochemistry theory and MCQs.
This is a shared conversation. Sign in to Orris to start your own chat.