Define and classify shock . Describe the pathogenesis and compensatory mechanisms activated in shock
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive, well-cited answer. Let me compose it.
Shock: Definition, Classification, Pathogenesis, and Compensatory Mechanisms
1. Definition
Shock is a state of systemic tissue hypoperfusion resulting from reduced cardiac output and/or reduced effective circulating blood volume, in which delivery of oxygen and nutrients is inadequate for normal cellular respiration. At the outset, the cellular injury is reversible; however, prolonged shock leads to irreversible tissue injury and is often fatal.
- Robbins & Kumar Basic Pathology: "Shock is a state in which diminished cardiac output or reduced effective circulating blood volume impairs tissue perfusion and leads to cellular hypoxia."
- Bailey and Love's Short Practice of Surgery, 28th ed.: "Shock is a systemic state of low tissue perfusion that is inadequate for normal cellular respiration. With insufficient delivery of oxygen and glucose, cells switch from aerobic to anaerobic metabolism."
2. Classification
The most clinically applicable classification is based on the initiating mechanism. All states share systemic tissue hypoperfusion, and different states may coexist in the same patient.
A. Hypovolemic Shock
- Due to an absolute reduction in circulating volume
- Causes: hemorrhage (hemorrhagic shock), vomiting, diarrhea, burns, third-spacing (bowel obstruction, pancreatitis), dehydration
- Mechanism: low cardiac output due to inadequate preload
- Most common form of shock; relative hypovolemia is a component of all other forms
B. Cardiogenic Shock
- Due to primary failure of the heart as a pump
- Causes: acute MI, ventricular rupture, arrhythmia, cardiomyopathy, blunt myocardial injury, valvular heart disease
- Extrinsic causes: cardiac tamponade, pulmonary embolism (sometimes classified separately as obstructive)
- Mechanism: intrinsic myocardial damage, extrinsic compression, or outflow obstruction
C. Obstructive Shock
- Due to mechanical obstruction of cardiac filling (reduced preload) despite normal myocardium
- Causes: cardiac tamponade, tension pneumothorax, massive pulmonary embolism, air embolism
- Mechanism: impaired filling of the left and/or right heart, leading to low cardiac output
D. Distributive Shock
- Due to abnormal distribution of blood flow - peripheral vasodilation with maldistribution of circulating volume
- Sub-types:
- Septic shock - most common distributive shock; overwhelming microbial infection (gram-positive bacteria > gram-negative > fungi); cytokine-mediated vasodilation + vascular leakage + DIC
- Anaphylactic shock - IgE-mediated hypersensitivity; massive systemic vasodilation + increased vascular permeability
- Neurogenic shock - spinal cord injury causing loss of vasomotor tone; acute vasodilation without tachycardia (bradycardia from loss of sympathetic tone)
E. Endocrine Shock
- Less commonly classified separately (Bailey & Love)
- Due to adrenal insufficiency (Addisonian crisis), hypothyroidism (myxedema crisis), or thyroid storm
- Mechanism: absence of cortisol sensitization of vessels to catecholamines
Table summary (from Robbins & Kumar Basic Pathology, Table 3.3):
| Type | Clinical Examples | Principal Mechanism |
|---|---|---|
| Cardiogenic | MI, tamponade, arrhythmia, PE | Pump failure - intrinsic myocardial damage, extrinsic compression, or outflow obstruction |
| Hypovolemic | Hemorrhage, burns, vomiting | Inadequate blood or plasma volume |
| Septic | Bacterial/fungal sepsis, toxic shock syndrome | Vasodilation, vascular pooling, endothelial activation/injury, DIC, cytokine cascade |
3. Pathogenesis
A. Cellular Level
When tissue perfusion falls, cells are deprived of oxygen and switch from aerobic to anaerobic metabolism. The product is lactic acid rather than CO2. Accumulation of lactate causes systemic metabolic acidosis.
As intracellular glucose is exhausted, anaerobic respiration ceases, leading to:
- Failure of the Na+/K+ ATPase pump
- Cellular swelling (water and Na+ entry)
- Release of lysosomal autodigestive enzymes
- Cell lysis and release of intracellular potassium (hyperkalaemia)
B. Microvascular Level
Progressive tissue ischaemia causes:
- Hypoxia and acidosis activate complement (C3a, C5a, C3b) and prime leukocytes
- Generation of oxygen free radicals and cytokine release (TNF, IL-1)
- Capillary endothelial injury - loss of tight junctions causes vascular leakage and tissue oedema, exacerbating hypoxia
- Activation of the coagulation cascade - microvascular thrombosis
C. Pathogenesis of Septic Shock (Detailed)
Septic shock, the most complex form, illustrates the full inflammatory cascade (Robbins, Cotran & Kumar Pathologic Basis of Disease):
1. Immune activation by microbial PAMPs:
- Microbial products (LPS, peptidoglycan, fungal glucans) engage pattern-recognition receptors on innate immune cells:
- Toll-like receptors (TLRs) recognizing PAMPs
- G-protein-coupled receptors (bacterial peptides)
- C-type lectin receptors (fungal components)
- Activated macrophages, neutrophils, and dendritic cells release: TNF, IL-1, IL-6, IL-12, IL-18, Type I interferons, ROS, prostaglandins
2. Proinflammatory state:
- Cytokines upregulate adhesion molecule expression on endothelium
- Complement cascade activated: C3a (mast cell activation), C5a (chemotaxis), C3b (opsonization)
- Widespread vascular leakage and tissue oedema impede nutrient delivery
3. Endothelial activation and injury:
- Cytokines loosen endothelial tight junctions → protein-rich oedema
- Activated endothelium produces nitric oxide (NO) and other vasoactive mediators
- NO causes vascular smooth muscle relaxation → systemic vasodilation and hypotension
4. Procoagulant state / DIC:
- Proinflammatory cytokines increase tissue factor production by monocytes and endothelial cells
- Decreased anticoagulant factors: tissue factor pathway inhibitor (TFPI), thrombomodulin, endothelial protein C receptor (EPCR)
- Increased PAI-1 dampens fibrinolysis
- Stasis from vascular leak amplifies coagulation factor activation
- Result: microvascular thrombosis (DIC) in up to 50% of septic patients, further compromising perfusion
5. Metabolic abnormalities:
- TNF, IL-1, and stress hormones cause insulin resistance and hyperglycemia
- Mitochondrial damage from oxidative stress impairs oxygen use even when delivered
6. Counterinflammatory immunosuppression:
- The initial hyperinflammatory phase triggers counterregulatory mechanisms
- Shift from Th1 (proinflammatory) to Th2 (anti-inflammatory) cytokines
- Production of soluble TNF receptor, IL-1 receptor antagonist, IL-10
- Lymphocyte apoptosis; patients oscillate between hyperinflammatory and immunosuppressed states
7. Organ dysfunction:
- Systemic hypotension + interstitial oedema + microvascular thrombosis → reduced O2/nutrient delivery
- Impaired mitochondrial function → failure to use delivered nutrients
- High cytokine levels diminish myocardial contractility
- Endothelial injury causes ARDS (Chapter 11 of Robbins)
- Multiorgan failure - kidneys, liver, lungs, heart
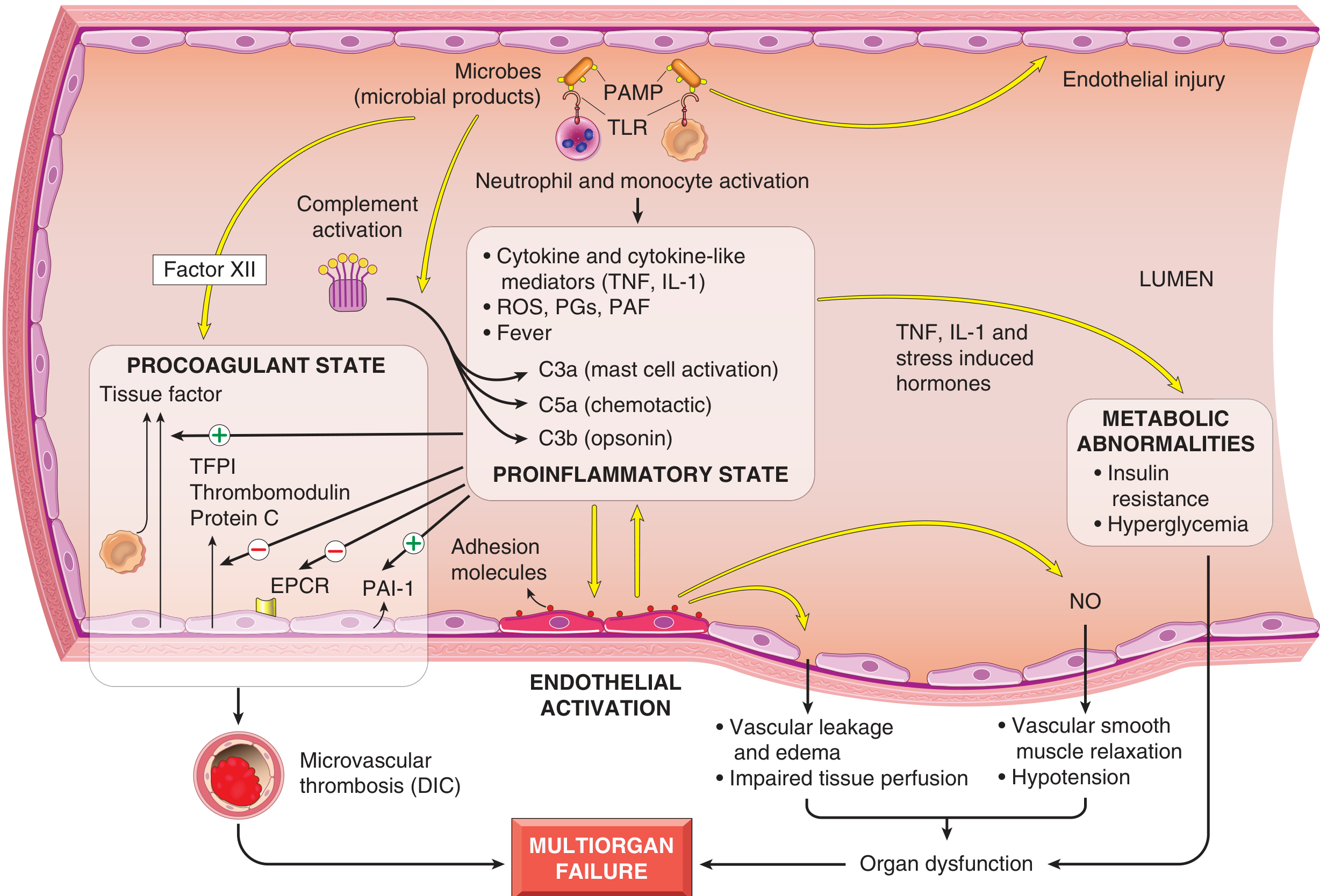
Fig: Major pathogenic pathways in septic shock. Microbial products activate endothelial cells and cellular/humoral elements of innate immunity, initiating cascades that produce a procoagulant, proinflammatory state leading to multiorgan failure. (Robbins & Kumar Basic Pathology)
4. Compensatory Mechanisms in Shock
Compensatory mechanisms are reflexes activated to maintain perfusion to vital organs (heart, brain) by sacrificing flow to non-essential organs (skin, muscle, gut). They are most clearly documented in hypovolemic shock but operate in all forms.
A. Immediate Neural Compensation (Seconds)
Baroreceptor reflex (most rapid):
- Reduced arterial pressure detected by baroreceptors in the carotid sinus and aortic arch
- Decreased stretch → decreased tonic inhibition of the vasomotor center in the medulla
- Result: increased sympathetic outflow to the heart and vasculature
Sympatho-adrenal response:
- Catecholamines (epinephrine + norepinephrine) released from adrenal medulla
- Effects:
- Tachycardia - increased heart rate and contractility (beta-1 receptors)
- Peripheral vasoconstriction - arteriolar constriction in skin, muscle, splanchnic beds (alpha-1 receptors)
- Venoconstriction - mobilizes venous reservoir, increases preload
- Cutaneous pallor - shunts blood to vital organs
- Coronary and cerebral vessels are relatively insensitive to sympathetic vasoconstriction and maintain flow
B. Hormonal Compensation (Minutes)
1. Renin-Angiotensin-Aldosterone System (RAAS):
- Decreased renal perfusion pressure activates juxtaglomerular cells → renin release
- Renin converts angiotensinogen → Angiotensin I → ACE converts to Angiotensin II
- Angiotensin II effects:
- Potent arteriolar vasoconstriction (raises BP)
- Stimulates aldosterone secretion from adrenal cortex
- Aldosterone → Na+ and water reabsorption in distal tubule → restored blood volume
2. ADH (Vasopressin):
- Released from posterior pituitary in response to decreased preload and hypovolemia
- Effects:
- Water reabsorption in renal collecting ducts → increased circulating volume
- At high concentrations (V1 receptors): vasoconstriction
3. Cortisol:
- Released from adrenal cortex in response to ACTH (stress response)
- Effects:
- Sensitizes blood vessels to catecholamines (permissive effect)
- Promotes sodium and water retention
- Gluconeogenesis to maintain fuel supply
C. Fluid Shifts (Minutes to Hours)
- Arteriolar constriction lowers capillary hydrostatic pressure
- Net movement of interstitial fluid into the intravascular space (transcapillary refill)
- Plasma volume partially restored ("autotransfusion")
D. Respiratory Compensation
- Metabolic acidosis and sympathetic stimulation increase respiratory rate and minute ventilation
- CO2 blown off → produces compensatory respiratory alkalosis to offset the metabolic acidosis
E. Renal Conservation
- Decreased GFR → reduced urine output (oliguria)
- RAAS activation → Na+ and water retention
- ADH → free water conservation
F. Hematological/Splenic Compensation
- Spleen contracts (in some species) → releases stored red blood cells
- Hemoconcentration temporarily increases oxygen-carrying capacity
5. Stages of Shock (Progression Despite Compensation)
(Robbins & Kumar; Guyton and Hall)
| Stage | Features |
|---|---|
| Nonprogressive (Compensated) | Compensatory mechanisms maintain perfusion to vital organs. Tachycardia, cool peripheries, maintained blood pressure. Occult anaerobic metabolism in skin/gut/muscle. |
| Progressive (Decompensated) | Tissue hypoperfusion widespread. Aerobic respiration replaced by anaerobic. Lactic acidosis worsens. Cellular injury in vital organs. Hypotension apparent. |
| Irreversible | Cellular and tissue injury so severe that even correcting hemodynamics cannot ensure survival. Multiorgan failure. Death. |
Key thresholds (Bailey & Love):
- Loss of ~15% circulating blood volume is within normal compensatory capacity
- Blood pressure typically falls only after 30-40% of circulating volume is lost
- Urine output below 0.5 mL/kg/hour signals failure of renal compensation
6. Morphological Changes (Organ Consequences)
When compensation fails, the following organ injuries occur:
- Brain: cerebral swelling, dysfunction, encephalopathy
- Heart: subendocardial ischaemia, myocardial dysfunction
- Lungs: ARDS - diffuse alveolar damage, hyaline membranes
- Kidneys: acute tubular necrosis (ATN) - most vulnerable to ischaemia
- Liver: centrilobular necrosis (zone 3 - furthest from blood supply)
- Gastrointestinal: mucosal ischaemia, bacterial translocation
Sources:
- Robbins & Kumar Basic Pathology, pp. 96-99 (Shock, Septic Shock, Stages of Shock)
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 133-137
- Bailey and Love's Short Practice of Surgery, 28th ed., Chapter 2 (Shock)
- Sabiston Textbook of Surgery (Physiology of Shock - Compensatory Mechanisms)
This is a shared conversation. Sign in to Orris to start your own chat.