Soft tissue sarcoma
Reading File
Finding Sources
Searching PubMed
"soft tissue sarcoma"[MeSH Terms] AND treatment
Reading File
Reading File
Searching PubMed
"sarcoma"[MeSH Terms] AND "treatment outcome"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the full answer.
Soft Tissue Sarcoma (STS)
Overview
Soft tissue sarcomas are a diverse group of approximately 70 distinct malignant neoplasms arising from mesenchymal tissues - skeletal muscle, adipose cells, blood and lymphatic vessels, and other connective tissue - as well as peripheral nerves (neuroectodermal origin). They are relatively rare, accounting for ~1% of cancer incidence and ~2% of cancer-related deaths in the United States (~13,400 new cases estimated annually).
The taxonomy of STS is extensive. The phylogeny below illustrates the major lineages:
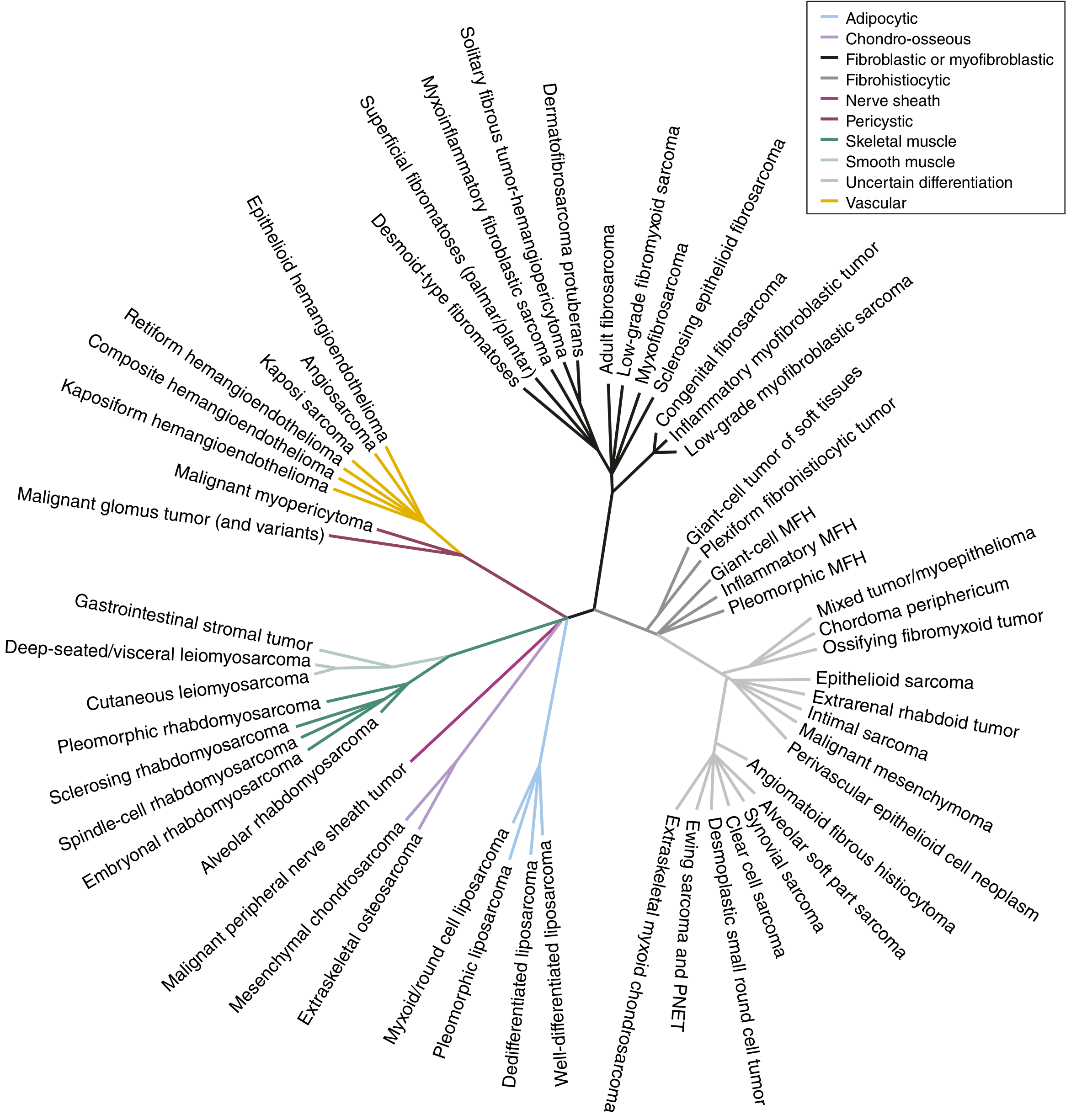
FIGURE 64.1 from Sabiston Textbook of Surgery: Unrooted phylogeny of ~60 STS subtypes, color-coded by lineage (adipocytic, smooth muscle, skeletal muscle, nerve sheath, vascular, etc.)
Epidemiology & Distribution
- Most common site: Thigh (44% of extremity STS), followed by other proximal limb locations
- Extremity > retroperitoneum > intraperitoneal in frequency
- Age linked to histologic subtype: Rhabdomyosarcoma, alveolar soft part sarcoma, and neurofibroma affect children/young adults; most adult STS is sporadic in middle-aged to older patients
- Wide clinical spectrum: from indolent low-grade neoplasms (e.g., well-differentiated liposarcoma) to highly aggressive tumors (e.g., angiosarcoma, desmoplastic small round cell tumor)
Risk Factors
Germline / Hereditary Syndromes
| Syndrome | Gene | Associated STS |
|---|---|---|
| Li-Fraumeni | TP53 (17p13.1) | Rhabdomyosarcoma, UPS, pleomorphic sarcoma |
| Neurofibromatosis type 1 (NF-1) | NF1 | Malignant peripheral nerve sheath tumor (MPNST) |
| FAP / Gardner syndrome | APC (5q21) | Desmoid tumors (esp. post-colectomy) |
| Hereditary retinoblastoma | RB1 (13q14) | Osteosarcoma, STS |
| Carney-Stratakis syndrome | SDHB/C/D | GIST + paraganglioma |
Environmental / Acquired
- Radiation exposure - post-radiation angiosarcoma; Stewart-Treves syndrome (lymphangiosarcoma in chronic lymphedema post-mastectomy)
- Chemical carcinogens (vinyl chloride, arsenic, dioxin) - hepatic angiosarcoma
Classification: Major Subtypes
1. Undifferentiated Pleomorphic Sarcoma (UPS)
- Previously called malignant fibrous histiocytoma (MFH)
- Location: Deep soft tissues of extremities, especially the thigh; middle-aged/older adults
- Histology: Sheets of large anaplastic polygonal cells with hyperchromatic bizarre nuclei; storiform (cartwheel) pattern of spindle and histiocytic cells around slit-like vessels; abundant atypical mitotic figures
- Complex karyotype from genomic instability
- Metastases in 17-50% of cases; generally poor prognosis
2. Liposarcoma
- Rarely arise in the subcutaneous tissues
- Hallmark: lipoblasts (signet ring-shaped cells); IHC: MDM2-positive
- Subtypes:
- Well-differentiated (low-grade, t(12;16) or 12q13-15 amplification)
- Dedifferentiated
- Myxoid/round cell - translocation t(12;16)(q13;p11) - FUS-DDIT3 fusion
- Pleomorphic (high-grade)
3. Rhabdomyosarcoma (RMS)
- Most common pediatric STS
- Alveolar subtype: translocation t(2;13) - PAX3-FOXO1 fusion
- Treatment: chemotherapy ± limb salvage surgery (unlike most other STS)
4. Synovial Sarcoma
- Common in young adults; foot and lower extremity
- Translocation t(X;18) - SYT-SSX fusion products
- One of the ESARC tumors (lymph node metastasis risk)
5. MPNST (Malignant Peripheral Nerve Sheath Tumor)
- NF-1 associated or de novo
- Histology: spindle cells in sweeping fascicles, whorled/nodular areas; S-100 positive on IHC
6. Leiomyosarcoma
- Histology: fascicular growth with spindle cells intersecting at right angles; IHC: SMA and desmin positive
7. Angiosarcoma
- Cells resemble vascular endothelium
- Highly malignant; lymph node and cutaneous metastases common
- Associated with Stewart-Treves syndrome
- May require amputation for local control
8. Epithelioid Sarcoma
- Most common STS of the hand; occurs in young adults
- Part of ESARC group (lymph node metastasis)
9. Clear Cell Sarcoma
- Common tumor of lower extremity/foot; young adults
- Lymph node metastasis risk (ESARC)
ESARC tumors (those with higher risk of lymph node metastases):
Epithelioid sarcoma, Synovial sarcoma, Angiosarcoma, Rhabdomyosarcoma, Clear cell sarcoma
Staging
Key prognostic factors:
| Factor | Adverse |
|---|---|
| Size | ≥5 cm |
| Grade | High grade |
| Depth | Deep to superficial fascia |
| Location | Retroperitoneal worse than extremity |
AJCC staging uses TNM + histologic grade (G1-3). High-grade, deep, >5 cm tumors are Stage III regardless of nodal status.
Diagnosis
Presentation
- Most common presentation: painless mass without prior evaluation
- Mimics include hypertrophic scar, myositis ossificans, hematoma, abscess, lipoma, cyst
- Red flags warranting workup: mass >5 cm, deep to fascia, fixed, growing
Imaging
- MRI - imaging modality of choice for trunk/extremity STS
- T1: decreased signal
- T2: increased signal
- Characterizes tumor relationship to neurovascular structures
- Chest CT - most frequent site of metastasis is the lung
- Additional imaging per subtype: abdomen/pelvis CT for myxoid liposarcoma, epithelioid sarcoma, leiomyosarcoma; paraspinal MRI for myxoid liposarcoma; brain imaging for alveolar soft part sarcoma, clear cell, angiosarcoma
Biopsy
- Core-needle biopsy is the standard approach (percutaneous, image-guided)
- Open incisional biopsy if core needle is non-diagnostic
- Biopsy tract must be oriented so it can be resected en bloc with the specimen
- Small (≤2 cm), superficial, mobile masses may be excised upfront with wide margins in selective cases
- Molecular/NGS testing is increasingly routine - changed diagnosis in 13% of patients in one series; FISH/RT-PCR for specific translocations
Treatment
Trunk and Extremity STS
Surgical principles:
- Wide local resection with negative margins is the cornerstone of treatment
- Limb salvage surgery (LSS) has replaced amputation as the standard; equivalent survival outcomes with better functional results
Radiation:
- Lesions >5 cm: LSS + radiation (XRT) - pre- or postoperative
- Lesions <5 cm, subcutaneous: LSS alone may suffice
- Preoperative radiation: lower total dose, smaller field, higher rate of wound complications
- Postoperative radiation: lower wound complication rate, but larger field and higher total dose
Chemotherapy:
- Standard first-line: doxorubicin ± ifosfamide
- Rhabdomyosarcoma: chemotherapy is a primary treatment modality (VAC regimen)
- Subtype-specific agents: trabectedin for leiomyosarcoma and myxoid liposarcoma; pazopanib for non-adipocytic STS; imatinib for GIST (KIT/PDGFRA mutation)
Retroperitoneal STS
- Surgery remains the cornerstone but complete resection is more challenging
- Higher local recurrence rates than extremity STS
- Role of preoperative radiation is being actively studied
Molecular Features Summary
| STS Subtype | Translocation / Mutation | Fusion Product |
|---|---|---|
| Synovial sarcoma | t(X;18) | SYT-SSX |
| Myxoid liposarcoma | t(12;16)(q13;p11) | FUS-DDIT3 |
| Alveolar rhabdomyosarcoma | t(2;13) | PAX3-FOXO1 |
| MPNST | NF1 loss | - |
| DFSP | t(17;22) | COL1A1-PDGFB |
| GIST | KIT / PDGFRA mutations | - |
| Well-diff. liposarcoma | 12q13-15 amplification | MDM2/CDK4 |
Recent Evidence (2024-2025)
Three notable recent RCTs are worth highlighting:
-
Pembrolizumab + radiation + surgery vs. radiation + surgery for Stage III extremity STS (SU2C-SARC032, Lancet 2024, PMID 39547252) - evaluating immunotherapy integration in localized disease
-
Doxorubicin-trabectedin + trabectedin maintenance vs. doxorubicin alone in leiomyosarcoma (NEJM 2024, PMID 39231341) - showed improved progression-free survival with combination therapy
Key Clinical Pearls (Miller's Orthopaedics)
- All STS subtypes share the same presentation, imaging, treatment, and differential - it is histology (and molecular signature) that distinguishes them
- ESARC = the 5 STS subtypes prone to lymph node metastasis
- NF-1 → MPNST; Stewart-Treves → angiosarcoma; FAP → desmoid
- Characteristic locations: Hand = epithelioid sarcoma; Foot = clear cell / synovial sarcoma
- Biopsy tract orientation is critical - must be excisable en bloc with the tumor
Sources:
- Sabiston Textbook of Surgery, 21st ed., Ch. 64 (Soft Tissue and Retroperitoneal Sarcoma)
- Miller's Review of Orthopaedics, 9th ed., Ch. 9 (Table 9.19, ESARC)
- Robbins, Cotran & Kumar Pathologic Basis of Disease (Undifferentiated Soft Tissue Sarcoma)
This is a shared conversation. Sign in to Orris to start your own chat.