Here is a comprehensive viva-ready review of Craniosynostosis compiled from Cummings Otolaryngology, Grainger & Allison's Diagnostic Radiology, Schwartz's Principles of Surgery, Bradley & Daroff's Neurology, and Creasy & Resnik's Maternal-Fetal Medicine.
CRANIOSYNOSTOSIS — Complete Neurosurgery Viva Guide
1. DEFINITION
Premature closure of one or more cranial sutures, leading to characteristic growth restriction perpendicular to the fused suture and compensatory growth parallel to it. The brain continues to grow, so the skull deforms predictably based on which suture is involved.
Virchow's Law: Growth is restricted perpendicular to the fused suture and compensated by expansion parallel to it.
2. DEMOGRAPHY & EPIDEMIOLOGY
| Parameter | Detail |
|---|
| Incidence | 1 in 2,000–2,500 live births |
| Sex | Males > Females (except coronal synostosis, where females predominate) |
| Age at presentation | Most identified at birth or within first few months of life |
| Most common type | Sagittal synostosis (40–55% of all cases) |
| Nonsyndromic : Syndromic ratio | ~80% : 20% |
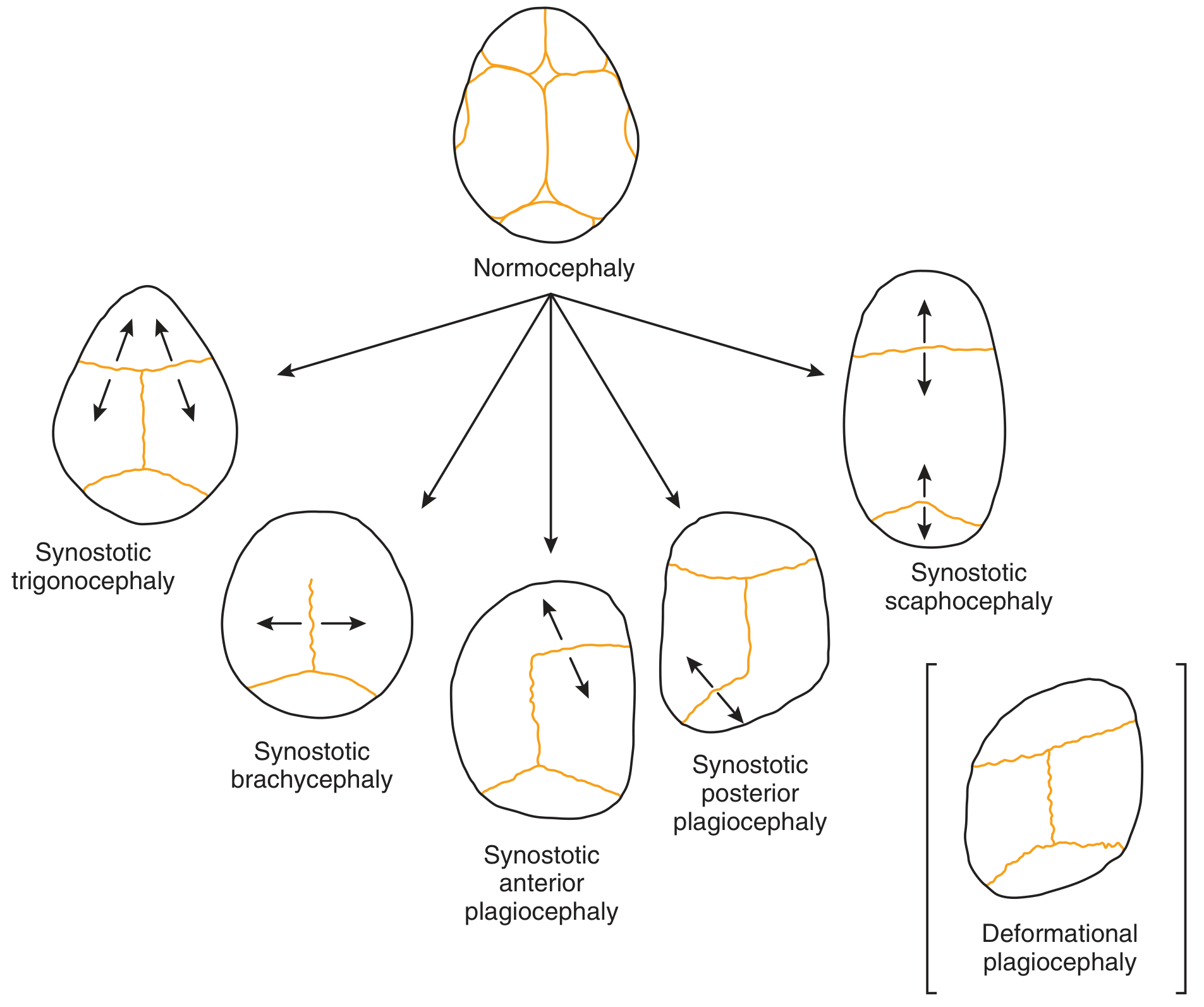
3. CLASSIFICATION
A. By Number of Sutures
- Simple (monosutural): 1 suture involved — most common
- Compound/Complex (multisuture): 2 or more sutures — usually syndromic
- Secondary: due to microcephaly, metabolic disease (rickets, hypophosphatasia), treated hydrocephalus, or drugs (valproate, phenytoin, aminopterin)
B. By Suture Involved
Fig. 187.1 — Compensatory growth directions for each suture type (Cummings Otolaryngology)
| Fused Suture | Skull Shape | Cephalic Index | Key Features |
|---|
| Sagittal (most common) | Scaphocephaly / dolichocephaly (boat-shaped) | < 75% (long, narrow) | Frontal bossing, occipital bullet |
| Metopic | Trigonocephaly (triangular, keel-shaped) | Reduced bifrontal diameter | Hypotelorism, midline ridge |
| Unilateral coronal | Anterior plagiocephaly | Asymmetric | Harlequin eye sign on XR, contralateral frontoparietal bossing |
| Bilateral coronal | Brachycephaly | > 90% (short, wide) | Sphenoid wing elevation, hypertelorism |
| Unilateral lambdoid (rarest) | Posterior plagiocephaly | Asymmetric | Must distinguish from deformational plagiocephaly (suture is open in the latter) |
| All sutures | Kleeblattschädel (cloverleaf skull) | Severe trilobed | Emergency surgery in neonatal period |
Cephalic Index (CI) = (biparietal diameter / fronto-occipital diameter) × 100
Normal: 75–85%
4. GENETICS & MOLECULAR PATHOLOGY
Key Genetic Mutations
| Gene | Condition | Inheritance |
|---|
| FGFR2 | Crouzon, Apert, Pfeiffer, Jackson-Weiss | AD |
| FGFR1 | Pfeiffer syndrome (some) | AD |
| FGFR3 | Muenke-type craniosynostosis | AD |
| TWIST1 | Saethre-Chotzen syndrome | AD |
| EFNB1 | Craniofrontonasal syndrome | X-linked |
| MSX2 | Boston-type craniosynostosis | AD |
Mechanism: FGFR mutations cause gain-of-function, leading to premature suture ossification via dysregulated osteoblast differentiation.
5. SYNDROMIC CRANIOSYNOSTOSIS — KEY SYNDROMES
Crouzon Syndrome
- Gene: FGFR2 (most common FGFR2 mutation → E253K or Y375C)
- Multi-suture: coronal, sagittal, metopic, squamosal
- Midface hypoplasia, hypertelorism, exorbitism (proptosis), dental malocclusion
- Normal limbs (distinguishes from Apert)
- Chiari I malformation common; hydrocephalus in 30%
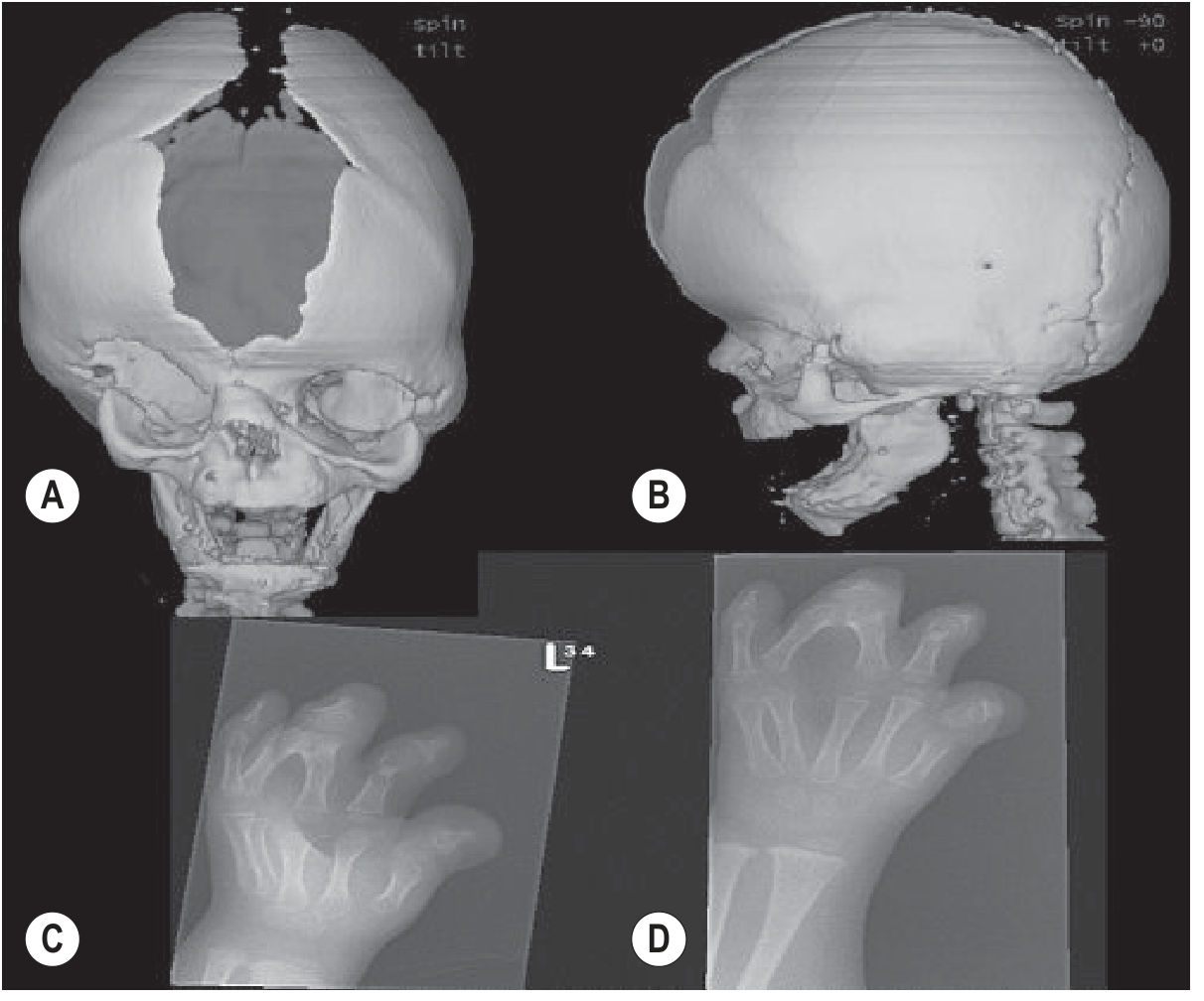
Apert Syndrome
- Gene: FGFR2 (Ser252Trp or Pro253Arg mutation)
- Bicoronal synostosis → brachycephaly
- Wide midline calvarial defect (sagittal + metopic region)
- Syndactyly of hands and feet — "mitten hand" or "hoof hand" (pathognomonic)
- Hypertelorism, midface hypoplasia, exorbitism
- Globe can sublux onto cheek in severe cases
Pfeiffer Syndrome
- Gene: FGFR1 or FGFR2
- Broad, medially deviated thumbs and big toes (brachydactyly)
- Types: I (mild), II (cloverleaf skull — most severe), III (severe without cloverleaf)
Saethre-Chotzen Syndrome
- Gene: TWIST1
- Coronal synostosis, low frontal hairline, ptosis, brachydactyly
Muenke Syndrome
- Gene: FGFR3 (Pro250Arg — most common FGFR mutation overall)
- Bilateral or unilateral coronal synostosis, sensorineural hearing loss
Fig. 76.52 — Apert syndrome: wide-open midline defect and syndactyly (Grainger & Allison's Diagnostic Radiology)
6. CLINICAL PRESENTATION
History
- Abnormal head shape noted at birth or within first few weeks/months
- Parents report hard ridge along suture line
- Functional symptoms (in severe/multisuture cases): irritability (raised ICP), visual changes, feeding difficulty, sleep apnoea (midface hypoplasia)
Examination Findings
- Palpable bony ridge over fused suture (pathognomonic)
- Absent fontanelle or prematurely closed fontanelle over affected suture
- Characteristic skull shape (see table above)
- Head circumference: Normal in single-suture; can be reduced in multisuture
- Neurological exam: Usually normal in isolated; fundoscopy for papilloedema (raised ICP)
- Ophthalmology: Proptosis, exposure keratopathy, strabismus (especially coronal)
- Hearing: Formal audiometry (especially Muenke/Saethre-Chotzen)
Features Suggesting Raised ICP
- Papilloedema
- Sunset sign of eyes
- Irritability, vomiting, bulging fontanelle
- Copper-beaten appearance on skull X-ray (chronic)
Neurodevelopmental Concerns
- Even single-suture synostosis carries higher risk of learning delays and neurodevelopmental problems vs. controls (Speltz et al. 2015)
- Unilateral coronal and lambdoid have highest risk; sagittal has lowest
- Trigonocephaly associated with language processing delays in severe cases
7. INVESTIGATIONS
A. Skull X-Ray (Plain Radiography)
Indications: Initial screening; widely available
Views: AP (coronal, sagittal, metopic), Towne (lambdoid, sagittal), Lateral (coronal, lambdoid)
Positive Findings:
- Absent or indistinct suture
- Bony bridging / sclerosis at fused suture
- Heaped-up / beaked suture margins
- Skull growth restriction shape (CI on lateral/AP)
- Harlequin eye sign (coronal) — elevated sphenoid wing makes orbit look like harlequin mask
- Copper-beaten skull — multiple gyral impressions indicating chronic ↑ ICP
- Note: A normal-looking suture does NOT exclude synostosis (fibrotic synostosis exists)
Key principle: Greater weight is given to skull shape than direct sutural appearance on plain film.
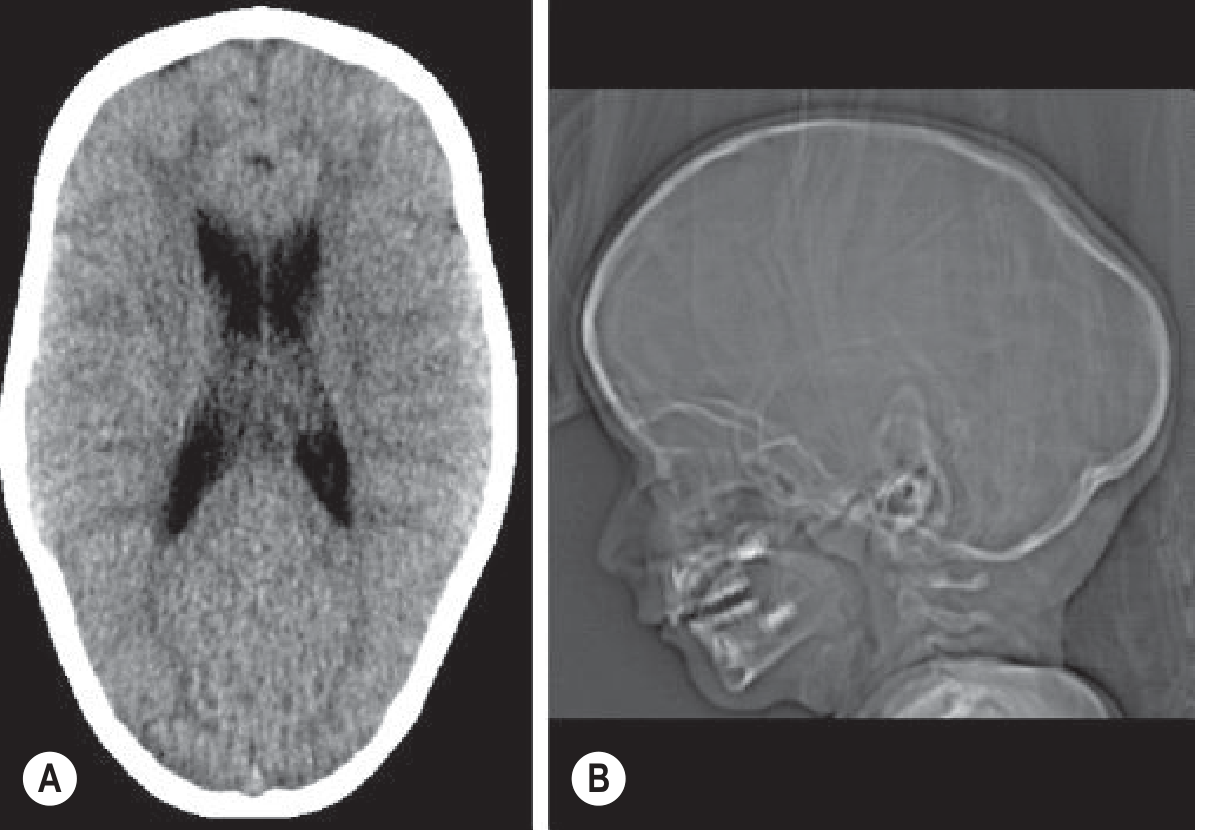
B. CT Scan (Gold Standard)
Indications: Confirm diagnosis; surgical planning; assess brain, skull base, venous sinuses
Protocol: Thin-slice (≤1 mm), low-mA, ± 3D reconstruction; CT venography if venous anatomy concern
Positive Findings:
- Sclerosis and bony bridging of suture (more sensitive than XR)
- 3D surface-shaded reconstruction — demonstrates skull deformity, suture status, and degree of deformity
- Harlequin deformity (coronal) — elevated sphenoid wing
- Hydrocephalus (most sensitive indicator of raised ICP — but detects only 40% of raised ICP cases)
- Chiari I malformation (especially Crouzon)
- Midline anomalies (callosal agenesis in Apert)
- Airway assessment — choanal atresia, nasopharyngeal stenosis
Fig. 76.50 — Sagittal synostosis: axial CT and lateral scout showing scaphocephaly (Grainger & Allison's)
C. MRI
Indications: Brain parenchymal assessment; Chiari malformation; posterior fossa; NOT first-line for suture assessment
Positive findings: Tonsillar herniation, callosal agenesis, ventriculomegaly, limbic abnormalities (Apert)
D. Ultrasound
Indications: Prenatal diagnosis (irregular/asymmetric calvarium shape on axial view); post-natal can confirm fused suture (no Doppler signal across suture)
- Fetal USS: irregular calvarium → suspect craniosynostosis; perform full anatomical survey
- Cloverleaf skull visible prenatally
E. Genetic Testing
Indications: Syndromic features, family history, multiple sutures, or ambiguous phenotype
- FGFR1/2/3 panel, TWIST1, EFNB1
- Important for recurrence risk counselling (AD conditions have 50% recurrence)
F. Invasive ICP Monitoring
Indications: Suspected raised ICP without clear imaging evidence; before repeat surgery in syndromic patients; papilloedema with inconclusive CT
- Epidural bolt or subdural catheter
- Normal ICP: <15 mmHg; >20 mmHg sustained = pathological raised ICP
G. Ophthalmological Assessment
Indications: All syndromic patients; any patient with visual complaints or papilloedema
- Visual evoked potentials (VEPs)
- Slit-lamp, fundoscopy, corneal exposure assessment
8. IMPORTANT SCORES / INDICES
| Score/Index | Formula | Normal | Significance |
|---|
| Cephalic Index (CI) | (BPD/FOD) × 100 | 75–85% | <75% = scaphocephaly; >90% = brachycephaly |
| Orbital Index | Orbital height/width × 100 | ~85% | Reduced in trigonocephaly |
| ICP (intracranial pressure) | Direct monitoring | <15 mmHg | >20 mmHg = indication for urgent surgery |
| Whitaker classification | Post-operative outcome | Categories I–IV | I = excellent to IV = major revision needed |
9. MANAGEMENT
Team Approach
A dedicated craniofacial team is mandatory: neurosurgeon, craniofacial plastic surgeon, ophthalmologist, orthodontist, neuropsychologist, speech therapist, audiologist, pediatric anesthesiologist.
When to Operate
| Situation | Timing |
|---|
| Kleeblattschädel | Within first weeks of life (emergency — prevents severe deficits/death) |
| Raised ICP, corneal exposure | Urgent — as early as feasible |
| Simple sagittal synostosis (endoscopic) | < 3 months (optimal for endoscopic strip craniectomy) |
| Open CVR (fronto-orbital advancement etc.) | 6–9 months (authors' preference) |
| Monobloc (forehead + orbits + midface) | 3–4 years |
| Whole orbit movements | 2–4 years |
| Occlusal correction | Teenage years |
Why early surgery? Infants <1 year have remarkable bony regenerative capacity, thinner/more pliable bone, and greater neuroplasticity. Early release may restore normal homeostasis. However, surgery too early risks growth inhibition (Fearon et al.: metopic repair at 4 months → more growth inhibition than at 12 months).
10. SURGICAL OPTIONS — DETAILED
A. Endoscopic Strip Craniectomy (ESC)
Indications: Single-suture synostosis (especially sagittal), age < 3–6 months, mild-moderate deformity
How:
- 1–2 small vertex incisions or a midline wavy incision
- Endoscope-assisted resection of the fused suture strip (2–3 cm wide)
- Followed by postoperative molding helmet for 3–6 months (critical — this is what reshapes the skull)
- No bone grafting, no bandeau
Advantages: Shorter operative time, less blood loss, shorter hospital stay, lower cost ($37,255 vs. $56,990 for open CVR), less transfusion requirement
Disadvantages: Requires postoperative helmet compliance; possible higher secondary surgery rate; endoscopic strip craniectomy alone (without helmet/springs) associated with poorer neurodevelopmental outcomes
Best results: Surgery at < 6 months correlates with best neurodevelopmental outcomes (best IQ, verbal scores, reading)
B. Open Cranial Vault Remodeling (CVR) / Total CVR (TCVR)
Indications: Single or multisuture; age 6–12 months; moderate-severe deformity; when endoscopic not feasible
Incision: Bicoronal (zigzag/wavy, postauricular extension)
How:
- Subgaleal/subperiosteal dissection
- Bone removed, reshaped/contoured, and replaced in corrected position
- Fixation: Resorbable plates and screws (preferred in infants — avoid titanium: risk of migration/growth interference; resorbable plates lose strength in 6 months, fully resorbed in <18 months)
- For sagittal: barrel-stave osteotomies to parietal bones ± biparietal wedge craniectomies ± frontal expansion
Barrel-stave osteotomy: Parallel cuts through outer cortex of parietal/temporal bone to allow it to spring outward; used to widen the skull in sagittal synostosis
C. Fronto-Orbital Advancement (FOA) / Fronto-Orbital Bar (FOB) Procedure
Indications: Metopic, unilateral, bilateral coronal synostosis — to address supraorbital retrusion, exorbitism, orbital malposition
Age: 6–9 months (open); can be done endoscopically
How:
- Bicoronal incision + subgaleal dissection
- Anterior pericranial flap elevated
- Supraorbital bandeau (bar of bone from orbit rims to above frontals) excised and reshaped
- Advanced anteriorly and fixed with resorbable plates
- Frontal bone also reshaped and advanced
D. Pi (π) Procedure
Indications: Sagittal synostosis — a subtype of open CVR
How: Pi-shaped osteotomy around fused sagittal suture, allowing biparietal widening and AP shortening. Can be done open or endoscopically.
E. Spring-Assisted Cranioplasty
Indications: Sagittal synostosis, age 3–6 months
How: Small incision; metallic springs placed across the osteotomy; spring force gradually expands the skull over 6 months; springs then removed (second procedure)
Advantage: Less invasive than full CVR; good for sagittal only
F. Distraction Osteogenesis (DO)
Indications: Syndromic multisuture; monobloc advancement; midface hypoplasia; posterior cranial vault expansion
Types:
- Posterior cranial vault distraction (PCVD): Increasingly used for multisuture syndromic; systematic review shows ↓ blood loss and ICP reduction
- Midface/Le Fort III distraction: For midface hypoplasia in Crouzon/Apert
Principle: Gradual bone regeneration in osteotomy gap at ~1 mm/day; allows larger advancement than acute osteotomy; reduced relapse
G. Monobloc Advancement
Indications: Combined forehead + orbital + midface correction — mainly syndromic (Crouzon, Apert)
Age: 3–4 years (can be earlier with DO)
Risk: Higher infection, CSF leak risk because intracranial space communicates with sinonasal cavity
H. Spring/Helmet Alone (Non-surgical)
Indications: Positional/deformational plagiocephaly (NOT true synostosis)
- Changing sleep position, physical therapy (torticollis), cranial molding orthosis (helmet)
- Well-demonstrated efficacy for positional plagiocephaly
11. ANESTHESIA CONSIDERATIONS (Viva Important)
- Small blood volume → blood transfusion commonly required (crossmatch preop)
- Impaired thermoregulation → keep room warm
- IV + arterial lines + bladder catheter (core temperature probe)
- Antibiotics + steroids in combined extracranial-intracranial procedures
- Eyes protected with ophthalmic ointment + clear plastic dressing; avoid corneal shields under bicoronal flap (pressure injury risk)
- Secure ETT firmly — head position changes during procedure
- Occipital deformities may require prone positioning
12. COMPLICATIONS OF SURGERY
| Complication | Notes |
|---|
| Blood loss / transfusion | Major risk, especially in young infants |
| Venous air embolism | Particularly when patient head-up |
| Infection | Higher in monobloc (open into sinuses) |
| CSF leak | Dural tear |
| Relapse to presurgical shape | More common with early surgery |
| Growth inhibition | From periosteal stripping and osteotomies |
| Raised ICP (persists/recurs) | Requires surveillance |
| Visual loss | From raised ICP or corneal exposure |
| Temporal hollowing / lateral orbital retrusion | Long-term — reported even at 5+ years follow-up |
| Idiopathic secondary stenosis | Re-fusion at index or new suture |
| Hardware migration (titanium) | Avoided by using resorbable plates |
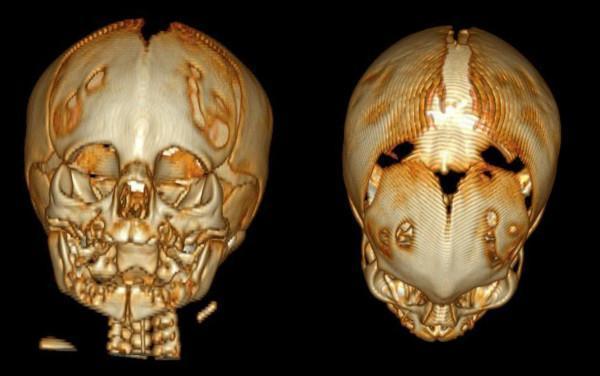
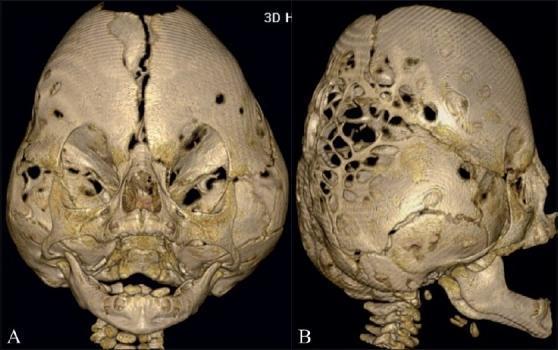
13. IMAGING GALLERY
Metopic Synostosis — 3D CT (trigonocephaly)
Kleeblattschädel (Cloverleaf Skull) — 3D CT
14. FOLLOW-UP
Frequency & Modality
| Phase | Frequency | Modality |
|---|
| Postoperative (first year) | 1 month, 3 months, 6 months, 12 months | Clinical exam, head circumference, CI measurement |
| Helmet therapy monitoring | Every 4–6 weeks until complete | Clinical + photographic documentation |
| Cranial growth surveillance (1–5 yrs) | 6-monthly | Clinical; XR if CI concern |
| ICP surveillance (syndromic/raised ICP) | Annual or as clinically indicated | Ophthalmology (fundoscopy/VEP), clinical exam; CT/MRI if deterioration |
| After syndromic repair | 6-monthly until skeletal maturity | Multidisciplinary: ophthalmology, neurosurgery, orthodontics, sleep study, audiology |
| Neurodevelopmental screening | 6 months, 12 months, 3 yrs, 5 yrs, school entry | Formal neuropsychological testing |
| Long-term (>5 years) | Annual | Look for temporal hollowing, lateral orbital retrusion, re-synostosis |
Key point: Hydrocephalus detects only 40% of children with raised ICP — clinical assessment (ophthalmology, symptom review) is essential at every visit.
15. VIVA POINTS — PEARLS
| Question | Answer |
|---|
| Most common type? | Sagittal → scaphocephaly |
| Rarest monosutural? | Unilateral lambdoid |
| What distinguishes positional from synostotic plagiocephaly? | In positional — suture is open on CT; head asymmetry without palpable ridge; treated with helmet/repositioning |
| Gene most commonly mutated in syndromic craniosynostosis? | FGFR2 |
| Most common FGFR mutation overall? | FGFR3 Pro250Arg → Muenke syndrome |
| What is Virchow's Law? | Growth restricted perpendicular to fused suture, compensated parallel to it |
| Harlequin sign — which suture? | Unilateral coronal — elevated sphenoid wing on skull XR |
| Copper-beaten skull — what does it mean? | Multiple gyral impressions from chronic raised ICP |
| Why resorbable plates preferred over titanium in infants? | Titanium can migrate/translocate intracranially with skull growth; resorbable plates lose strength in 6 months, resorb in <18 months |
| Endoscopic vs. open CVR — key difference? | Endoscopic: less blood loss, shorter stay, needs helmet; must be done < 3–6 months |
| In what syndrome is globe subluxation onto cheek possible? | Apert syndrome |
| What distinguishes Crouzon from Apert? | Crouzon has normal limbs; Apert has syndactyly |
| Chiari I more common in which syndrome? | Crouzon (more skull base sutural synostosis) |
| Best age for sagittal synostosis endoscopic surgery for neurodevelopment? | < 6 months |
| What score/index measures head shape? | Cephalic Index = (BPD/FOD) × 100; normal 75–85% |
Sources: Cummings Otolaryngology Head & Neck Surgery (Ch. 187); Grainger & Allison's Diagnostic Radiology (Ch. 76); Schwartz's Principles of Surgery (11th ed.); Bradley & Daroff's Neurology in Clinical Practice; Creasy & Resnik's Maternal-Fetal Medicine.
Recent evidence note: A 2023 systematic review (PMID 37052891) on posterior cranial distraction supports its use in syndromic multisuture craniosynostosis with favorable ICP outcomes. A 2023 systematic review on idiopathic secondary stenosis (PMID 37316986) highlights re-synostosis as an important long-term complication to monitor.