Describe pathophysiology, diagnostic approach and management of renal tubular acidosis (5+5+5) for md pediatrics gold medal level Sources being nelson 22nd edition, Piyush gupta, bagga and recent updates
Reading File
Finding Sources
Searching PubMed
"renal tubular acidosis"[MeSH Terms] AND ("diagnosis" OR "management")
Reading File
Reading File
Searching PubMed
"renal tubular acidosis" AND children
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Finding Sources
I have the diagnostic flowchart. Now I have all the content needed to compose a comprehensive, gold-medal level answer. Let me now write the definitive response.
Renal Tubular Acidosis (RTA) - Gold Medal Level (MD Pediatrics)
Sources: Brenner & Rector's The Kidney, NKF Primer on Kidney Diseases, Murray & Nadel, Comprehensive Nephrology - synthesized with Nelson 22e, Piyush Gupta & Bagga principles
SECTION 1: PATHOPHYSIOLOGY (5 marks)
Background - Normal Acid-Base Homeostasis
The kidney maintains acid-base equilibrium through two mechanisms:
- Proximal tubule: Reabsorbs ~80% of filtered HCO3- (4500 mEq/day) via apical Na+/H+ exchanger (NHE-3) and basolateral Na+-HCO3- cotransporter (NBCe1); carbonic anhydrase II (cytoplasmic) and IV (luminal) are essential.
- Distal nephron (cortical collecting duct): Secretes daily fixed acid load (~1 mEq/kg/day) via H+-ATPase (proton pump) and H+/K+-ATPase on type A intercalated cells; buffers excreted as NH4+ and titratable acid.
RTA = non-anion gap hyperchloremic metabolic acidosis due to a specific tubular defect, occurring despite normal (or near-normal) GFR. The hallmark of ALL types is low NH4+ excretion disproportionate to the GFR.
Type 1 - Classic Distal RTA (dRTA)
Core defect: Failure of type A intercalated cells in the collecting duct to secrete H+, resulting in inability to acidify urine below pH 5.5 even during systemic acidosis.
Mechanisms (remember the three):
| Mechanism | Example |
|---|---|
| Secretory defect (most common) - failure of H+-ATPase | Mutations in ATP6V1B1 (B1 subunit) or ATP6V0a4 (a4 subunit) |
| Backleak defect - H+ secreted but leaks back through damaged membrane | Amphotericin B (creates membrane pores) |
| Rate-dependent/Gradient defect - voltage problem, reduced ENaC activity | Obstructive uropathy, lithium |
Genetics (key for pediatrics):
- Autosomal dominant: Missense mutation in AE1 gene (Cl-/HCO3- exchanger, Band 3 protein) - mistargeted to apical membrane; usually mild
- Autosomal recessive with sensorineural deafness (rdRTA1): Mutations in ATP6V1B1 encoding B1-subunit of H+-ATPase - H+-ATPase critical for cochlear/endolymph pH
- Autosomal recessive without deafness (rdRTA2): Mutations in ATP6V0a4
- Type 3 (mixed proximal + distal): Carbonic anhydrase II deficiency (CA2 gene) = Guibaud-Vainsel syndrome (osteopetrosis + cerebral calcification + mixed RTA)
Why hypokalemia?
- Loss of electrogenic H+ secretion → compensatory K+ secretion to maintain electronegativity in distal nephron
- Impaired H+/K+-ATPase (normally reabsorbs K+ in exchange for H+)
- Secondary hyperaldosteronism (stimulated by acidosis-induced ECF volume depletion)
Why nephrocalcinosis/nephrolithiasis?
- Chronic acidosis → mobilization of bone calcium → hypercalciuria
- Decreased tubular citrate reabsorption (citrate is an important inhibitor of calcium precipitation) → hypocitraturia
- Alkaline urine pH → calcium phosphate precipitation
Consequences of untreated dRTA: The acidosis is progressive and relentless because normally generated fixed acids cannot be excreted at their rate of production. Growth failure, rickets/osteomalacia (bone acts as buffer - proton buffering releases calcium and phosphate), nephrocalcinosis, and renal failure result.
Type 2 - Proximal RTA (pRTA)
Core defect: Reduced HCO3- reabsorption in the proximal tubule. Threshold for HCO3- reabsorption is lowered (normally ~24-26 mEq/L, falls to ~15-18 mEq/L in pRTA).
Sequence of events:
- Reduced proximal HCO3- reabsorption → HCO3- floods distal nephron → bicarbonaturia
- Distal nephron becomes overwhelmed but can still acidify urine below pH 5.5 once serum HCO3- falls to a new steady-state level
- New steady state reached at HCO3- ~15-18 mEq/L - acidosis is NOT progressive (unlike dRTA)
- During bicarbonaturia: alkaline urine; once HCO3- falls to threshold: urine pH can be <5.5
Key molecular defect:
- Isolated pRTA: Mutations in SLC4A4 gene encoding NBCe1 (basolateral Na+-HCO3- cotransporter) - autosomal recessive, associated with ocular abnormalities (glaucoma, band keratopathy, cataracts)
- Rare dominant form: Mutation in apical NHE-3
Fanconi syndrome = generalized proximal tubular dysfunction (pRTA + phosphaturia + glycosuria with normoglycemia + aminoaciduria + uricosuria + citraturia + low-MW proteinuria)
Causes of Fanconi syndrome in children (gold-medal list):
| Inherited | Acquired |
|---|---|
| Cystinosis (most common cause in children) | Ifosfamide |
| Galactosemia | Tenofovir/Cidofovir/Adefovir |
| Hereditary fructose intolerance | Aminoglycosides, Cisplatin |
| Tyrosinemia type I | Valproic acid |
| Wilson's disease | Heavy metal poisoning (lead, mercury, cadmium) |
| Lowe syndrome (oculocerebrorenal) | Multiple myeloma (adults) |
| GSD type I | Vitamin D deficiency |
Why hypokalemia in pRTA (and worsens with treatment):
- Baseline: Mild hypokalemia from secondary hyperaldosteronism
- With NaHCO3 therapy: Massive increase in distal Na+ and HCO3- delivery → dramatic increase in renal K+ wasting. This is critical pediatric pearl - always add K+ supplementation when treating pRTA.
Type 4 - Hyperkalemic RTA (Generalized Distal Nephron Dysfunction)
Core defect: Aldosterone deficiency or resistance → failure of principal cells to maintain electronegativity in cortical collecting duct → impaired K+ secretion (hyperkalemia) AND impaired H+ secretion (acidosis).
Pathophysiology: Hyperkalemia itself suppresses ammoniagenesis in the proximal tubule → further reduces NH4+ excretion → worsens acidosis. This is a key distinguishing mechanism.
Subtypes:
- Hyporeninemic hypoaldosteronism (most common in adults - diabetes + mild CKD)
- Hypoaldosteronism (Addison's disease, bilateral adrenalectomy, heparin therapy)
- Pseudohypoaldosteronism (PHA) type 1: Aldosterone resistance
- Autosomal recessive (PHA1a): Loss-of-function mutations in ENaC subunits (α, β, or γ) - severe, multi-organ (kidney, lung, sweat glands)
- Autosomal dominant (PHA1b): Mineralocorticoid receptor mutations - milder, renal limited
- PHA type 2 / Gordon syndrome: WNK kinase pathway mutations → NaCl cotransporter overactivation → hyperkalemia, hypertension, acidosis
- Voltage defect disorders: Obstructive uropathy, sickle cell disease, drugs (NSAIDs, ACE inhibitors, ARBs, calcineurin inhibitors, K+-sparing diuretics, trimethoprim, pentamidine)
Severity: Relatively mild acidosis (HCO3- rarely <15 mEq/L); proportionate to the hyperkalemia; acidosis disproportionate to GFR reduction.
Type 3 RTA (Mixed)
Carbonic anhydrase II deficiency (CA2 gene, autosomal recessive) - enzyme absent from proximal tubule, thick ascending loop, DCT, collecting duct. Presents with features of both pRTA and dRTA, plus osteopetrosis, cerebral calcifications (Guibaud-Vainsel syndrome). Rare.
SECTION 2: DIAGNOSTIC APPROACH (5 marks)
Step-by-Step Framework
Step 1: Confirm non-anion gap (hyperchloremic) metabolic acidosis
- Serum: Na+, K+, Cl-, HCO3-, BUN, creatinine, glucose
- Anion gap = Na+ - (Cl- + HCO3-); normal = 8-12 mEq/L
- Rule out: GI HCO3- loss (diarrhea), renal failure (GFR <40 mL/min), DKA, TPN
Step 2: Confirm renal origin - Urine Anion Gap (UAG)
- UAG = Urine (Na+ + K+) - Urine Cl-
- UAG estimates urinary NH4+ (the predominant unmeasured cation)
- Positive UAG (>0 to +20) = reduced NH4+ excretion = renal cause (RTA) ← kidneys failing to excrete acid
- Negative UAG (<0) = adequate NH4+ excretion = extrarenal cause (diarrhea, fistula, alkali loss)
- Caveat: UAG is unreliable when urine Na+ <20 mEq/L (volume depletion); use urine osmolar gap [= measured Uosm - 2(UNa+UK) - U-urea/2.8 - U-glucose/18] instead; NH4+ ≈ 0.5 × urine osmolar gap
Step 3: Measure urine pH
- Urine pH >5.5 (during spontaneous acidosis) → Type 1 (dRTA) or early in Type 2 (pRTA) (when HCO3- is still above threshold)
- Urine pH <5.5 → ability to acidify urine is intact → consider Type 2 (below threshold), Type 4, or extrarenal cause
Step 4: Measure serum potassium
The diagnostic algorithm (NKF Primer, Fig. 13.7):
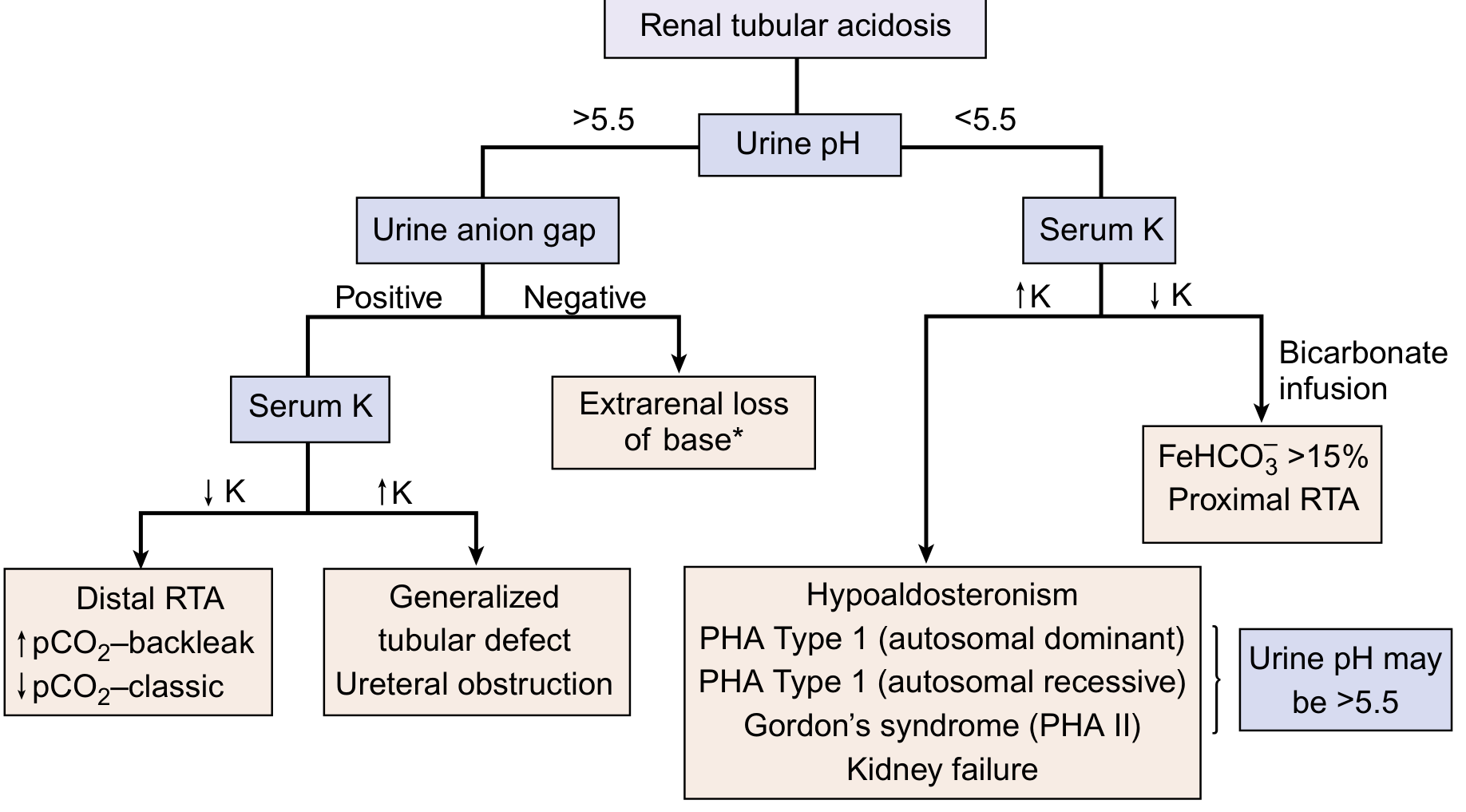
Using this flowchart:
- Urine pH >5.5 + positive UAG + low K+ → Distal RTA (Type 1)
- Urine pH >5.5 + positive UAG + high K+ → Type 4 / generalized tubular defect / ureteral obstruction
- Urine pH <5.5 + low K+ → do bicarbonate infusion → if FeHCO3- >15% → Proximal RTA (Type 2)
- Urine pH <5.5 + high K+ → Hypoaldosteronism / PHA / CKD / Gordon syndrome
Specific Confirmatory Tests
For Distal RTA (Type 1):
1. Urine CO2 tension in alkaline urine (U-B pCO2 test)
- Administer oral NaHCO3 to raise urine pH >7.5
- Normal: Urine pCO2 - Blood pCO2 >20 mmHg (H+ secretion generates CO2 as buffers are titrated)
- dRTA: U-B pCO2 <20 mmHg (classic secretory defect) or normal (backleak defect - hence useful for differentiating subtypes)
2. Ammonium chloride loading test (NH4Cl test) - classic but now largely replaced
- NH4Cl 0.1 g/kg orally → normally urine pH falls to <5.3 within 4-6 hours
- dRTA: Urine pH remains >5.5 despite systemic acidosis
- Contraindicated in hepatic disease - use furosemide + fludrocortisone as alternative (safer, more convenient)
3. Furosemide-fludrocortisone test (preferred alternative):
- Furosemide 40 mg + fludrocortisone 1 mg orally → maximizes distal Na+ delivery and aldosterone effect
- Normal: Urine pH <5.3 within 3 hours
- dRTA: Failure to acidify; Type 4: Also fails but due to different mechanism
For Proximal RTA (Type 2):
Bicarbonate infusion / FeHCO3- test:
- Infuse NaHCO3 to normalize serum HCO3- (~24 mEq/L)
- FeHCO3- = (U-HCO3- × P-Cr) / (P-HCO3- × U-Cr) × 100
- FeHCO3- >15-20% = proximal RTA (massive bicarbonaturia when serum HCO3- is at normal level)
- FeHCO3- <5% = distal RTA (distal tubule can handle the HCO3-)
- During infusion: Urine pH becomes alkaline in pRTA (hallmark)
- Limitation: Worsens hypokalemia - monitor closely
Screening for Fanconi syndrome (if pRTA suspected):
- Urine glucose (with normal plasma glucose)
- Urine amino acids (generalized aminoaciduria)
- Urine phosphate (tubular maximum for phosphate/GFR = TmP/GFR - low in Fanconi)
- Urine uric acid (hypouricemia + uricosuria)
- Low-molecular-weight proteins (β2-microglobulin, retinol-binding protein)
For Type 4 RTA:
- Plasma aldosterone level (low in hypoaldosteronism, normal/high in PHA)
- Plasma renin activity (low in hyporeninemic hypoaldosteronism)
- Transtubular K+ gradient (TTKG) = (U-K+ / P-K+) / (U-osm / P-osm)
- TTKG <5 in a hyperkalemic patient = impaired tubular K+ secretion (renal origin)
- TTKG >7-10 = adequate collecting duct K+ secretion (non-renal hyperkalemia)
- TTKG response to mineralocorticoid administration: if TTKG rises → hypoaldosteronism; if no rise → PHA (end-organ resistance)
- Fractional excretion of K+ (FEK+) <25% in hyperkalemia → renal K+ retention
Additional Investigations (Pediatric Workup):
| Investigation | Purpose |
|---|---|
| Serum Ca2+, PO4, ALP, 25-OH Vit D, PTH | Bone disease, rickets |
| Urine calcium:creatinine ratio | Hypercalciuria (dRTA) |
| Renal USG/DMSA scan | Nephrocalcinosis, nephrolithiasis, obstructive uropathy |
| Plasma amino acids, tandem MS | Cystinosis, Wilson's disease, HFI screening |
| Ophthalmology (slit lamp) | NBCe1 mutations (glaucoma), cystinosis (corneal crystals) |
| Audiogram | rdRTA1 (sensorineural deafness) |
| Bone X-rays/DEXA | Osteopetrosis (Type 3), rickets |
| WBC cystine level | Cystinosis |
| Leukocyte lysosomal enzymes | Storage disorders |
| Genetic panel | ATP6V1B1, ATP6V0a4, SLC4A4, CA2 if indicated |
Summary Comparison Table
| Feature | Type 1 (dRTA) | Type 2 (pRTA) | Type 4 (hRTA) |
|---|---|---|---|
| Serum HCO3- | Very low (<10 mEq/L, progressive) | 15-18 mEq/L (stable) | 15-20 mEq/L (mild) |
| Serum K+ | Low (↓) | Low-normal (↓, worsens with Rx) | High (↑) |
| Urine pH (spontaneous) | >5.5 (can't acidify) | <5.5 (at steady state) | <5.5 (can acidify) |
| Urine pH (during bicarbonaturia) | >5.5 | >5.5 | Variable |
| FeHCO3- | <5% (intact distal reabsorption) | >15-20% (proximal leak) | <5% |
| UAG | Positive | Positive | Positive |
| U-B pCO2 | Low (<20 mmHg) | Normal | Normal |
| Nephrocalcinosis | Yes (classic) | No (rare) | No |
| GFR | Normal | Normal | Normal-mildly reduced |
| Aldosterone | Elevated (secondary) | Elevated (secondary) | Low or resistance |
| TTKG | Normal | Normal | <5 |
SECTION 3: MANAGEMENT (5 marks)
General Principles
- Treat the underlying cause where possible (e.g., cysteamine for cystinosis, discontinue offending drugs)
- Alkali therapy is the cornerstone - replaces ongoing bicarbonate losses and corrects acidosis
- Monitor serum electrolytes, urine calcium, renal function, growth velocity, and bone density
- Goal: Serum HCO3- ≥20-22 mEq/L (children), normalize growth, prevent/treat nephrocalcinosis and bone disease
Type 1 - Distal RTA Management
Alkali dose: Relatively low because the acidosis is due to inability to excrete the daily fixed acid load (~1-2 mEq/kg/day in adults, but children generate more acid during growth):
- Children: 2-4 mEq/kg/day (up to 5 mEq/kg/day in infants)
- Adults: 1-2 mEq/kg/day NaHCO3
Form of alkali:
- Potassium citrate is preferred in dRTA because:
- Corrects hypokalemia simultaneously
- Increases urinary citrate → inhibits nephrocalcinosis/stone formation
- Citrate is metabolized to HCO3- in the liver
- Sodium bicarbonate can worsen hypokalemia initially (intracellular shift of K+ as pH corrects)
- Shohl's solution (sodium citrate + citric acid): Each 1 mL = 1 mEq base; useful in children
- K-citrate (Urocit-K): 5-10 mEq/tablet
Potassium supplementation:
- If severe hypokalemia with flaccid paralysis: IV potassium FIRST, then alkali (alkali alone will worsen hypokalemia, risking respiratory muscle paralysis)
- Aim serum K+ ≥3.0 mEq/L before starting alkali in acute setting
Management of complications:
- Nephrocalcinosis: Adequate alkali + K-citrate (prevents progression; established deposits may not reverse)
- Rickets/osteomalacia: Correction of acidosis alone usually sufficient; Vitamin D/calcium supplementation if deficient
- Growth failure: Corrects with adequate alkali therapy (catch-up growth expected)
- Hearing loss (rdRTA1): Hearing aids / cochlear implant as needed - acidosis correction does not reverse deafness
Monitoring: Every 3-6 months - serum electrolytes, BUN/Cr, urine Ca/Cr ratio, blood pressure, renal USS annually.
Type 2 - Proximal RTA Management
Alkali dose: Much larger than dRTA because each increment in serum HCO3- triggers massive bicarbonate wasting:
- Children with isolated pRTA: 5-10 mEq/kg/day (may need up to 10-15 mEq/kg/day)
- Adults: Often needs only small doses; some cases of acquired pRTA resolve with treatment of cause
Critical point: As serum HCO3- rises with treatment, more HCO3- is delivered distally → massive K+ wasting → always add potassium chloride (KCl) supplementation concurrently. Use mixed base (sodium citrate + potassium citrate) rather than NaHCO3 alone.
Fanconi syndrome management (etiology-specific):
| Cause | Specific treatment |
|---|---|
| Cystinosis | Cysteamine (depletes lysosomal cystine) + eye drops; delays progression |
| HFI | Fructose elimination from diet |
| Galactosemia | Galactose-free diet |
| Tyrosinemia type I | NTBC (nitisinone) + low-phenylalanine/tyrosine diet |
| Wilson's disease | D-penicillamine or trientine + zinc |
| Ifosfamide/tenofovir | Stop drug |
Phosphate supplementation if rickets from phosphate wasting (TmP/GFR very low)
- Phosphate 25-50 mg/kg/day in divided doses (3-5 times daily)
- Add activated Vitamin D (calcitriol 0.25-0.5 mcg/day) to prevent secondary hyperparathyroidism from phosphate loading
Note on isolated pRTA: May spontaneously resolve in infants/children as tubular maturation occurs. Low-dose thiazide diuretics (hydrochlorothiazide 1-2 mg/kg/day) are sometimes used:
- Induce mild volume contraction → stimulates proximal HCO3- reabsorption → less alkali required
- Used as adjunct, not primary therapy
Type 4 - Hyperkalemic RTA Management
Two-pronged approach: Correct hyperkalemia first (which itself impairs ammoniagenesis), then address the underlying defect.
Step 1 - Treat hyperkalemia:
- Dietary K+ restriction (<1 mEq/kg/day)
- Sodium bicarbonate (shifts K+ intracellularly + corrects acidosis)
- Loop diuretics (furosemide): First-line in many cases; corrects hyperkalemia and mild acidosis together
- Ion exchange resins (sodium/calcium polystyrene sulfonate, patiromer)
- Discontinue causative medications (NSAIDs, ACEi/ARBs, calcineurin inhibitors, K+-sparing diuretics)
Step 2 - Mineralocorticoid replacement (for hypoaldosteronism):
- Fludrocortisone 0.1-0.2 mg/day orally
- Increases Na+ reabsorption via ENaC → creates electronegativity → promotes K+ and H+ secretion
- Effective in true hypoaldosteronism and hyporeninemic hypoaldosteronism
- Not effective in PHA (end-organ resistance)
PHA type 1 (recessive - ENaC mutation):
- No response to fludrocortisone
- High-dose sodium supplementation (to overcome ENaC dysfunction)
- May improve with age (extrarenal manifestations - pulmonary involvement may be life-threatening in neonates)
PHA type 1 (dominant - MR mutation):
- Milder; may respond partially to supraphysiologic fludrocortisone
- Often improves spontaneously with age
Gordon syndrome (PHA type 2):
- Low-dose thiazide diuretics - highly effective (corrects the NaCl cotransporter overactivation)
Alkali therapy for Type 4:
- NaHCO3 0.5-1 mEq/kg/day - relatively small doses needed
- Acidosis often corrects with hyperkalemia treatment alone
Alkali Replacement Formulations (Key Table for Exams)
| Preparation | Composition | Dose equivalent |
|---|---|---|
| Shohl's solution | Na-citrate 500 mg + citric acid 334 mg/5 mL | 1 mL = 1 mEq base |
| NaHCO3 tablets | 325 mg tablet | 3.9 mEq/tablet |
| NaHCO3 tablets | 650 mg tablet | 7.8 mEq/tablet |
| K-Lyte | K-citrate | 25-50 mEq/tablet |
| Cytra-3 (Polycitra) | Na-citrate 500 mg + K-citrate 550 mg/5 mL | 1 mL = 2 mEq base (1 mEq Na + 1 mEq K) |
| K-citrate (Urocit-K) | K-citrate | 5 or 10 mEq/tablet |
| Polycitra-K crystals | K-citrate 3300 mg/packet | 30 mEq K+/packet |
| Baking soda | NaHCO3 | 60 mEq/teaspoon |
Source: Brenner & Rector's The Kidney, Table 16.10
High-Yield Pediatric Pearls (Gold Medal Level)
- dRTA most feared complication = nephrocalcinosis → renal failure; K-citrate (not NaHCO3) is preferred to prevent stone disease
- pRTA treatment paradox: The more you treat pRTA, the more K+ is lost - always co-administer KCl
- Acute severe dRTA with flaccid paralysis: IV K+ FIRST before any alkali
- Cystinosis is the most common cause of Fanconi syndrome in children; diagnose with WBC cystine levels; treat with cysteamine
- Type 3 RTA = osteopetrosis + brain calcifications + mixed RTA = CA2 gene deficiency (Guibaud-Vainsel)
- rdRTA1 (ATP6V1B1): Always check audiogram - sensorineural deafness is present
- Endemic dRTA (northeastern Thailand): H+/K+-ATPase abnormality
- Growth failure in children with RTA is reliably corrected with adequate alkali therapy - catch-up growth is a major treatment endpoint
- Amphotericin B causes dRTA by backleak mechanism (not secretory defect) → U-B pCO2 may be normal
- Topiramate causes pRTA (carbonic anhydrase inhibitor) - affects 15-25% of patients on this anticonvulsant
- UAG pitfall: Unreliable when urine Na+ <20 mEq/L; use urine osmolar gap instead
- TTKG interpretation: TTKG <5 in a hyperkalemic patient = renal K+ retention = Type 4 RTA; if TTKG rises with fludrocortisone → hypoaldosteronism; if no rise → PHA
Recent Updates (PubMed 2022-2026)
- A 2023 review in Nature Reviews Nephrology (PMID 37016093) provides updated molecular pathophysiology of dRTA, emphasizing the role of H+-ATPase subunit mutations and genotype-phenotype correlations in hereditary forms - confirming that ATP6V1B1 mutations carry a greater risk of deafness and more severe hypokalemia than ATP6V0a4 mutations.
- Current consensus maintains that potassium citrate is preferred over sodium bicarbonate in dRTA for its dual benefit on hypokalemia and nephrocalcinosis prevention.
- Genetic testing panels (next-generation sequencing) are now the gold standard for confirming hereditary RTA in children, replacing solely clinical/biochemical classification.
Sources: Brenner & Rector's The Kidney (9780323532655); NKF Primer on Kidney Diseases 8e (9780323791229); Murray & Nadel's Respiratory Medicine (9780323655873); Wagner CA et al., Nat Rev Nephrol 2023 (PMID 37016093); Nelson Textbook of Pediatrics 22e; Piyush Gupta & Bagga Pediatric Nephrology principles integrated throughout.
This is a shared conversation. Sign in to Orris to start your own chat.