explain
cd /home/daytona/workspace/attachments/37076cbd-db80-4cef-98d8-c35834427ad8/ && …
cd /home/daytona/workspace/attachments/37076cbd-db80-4cef-98d8-c35834427ad8/ && …
Emphysema - Complete Explanation
1. What is COPD?
- Currently the 4th leading cause of death worldwide
- Emphysema and chronic bronchitis are the two major forms of COPD - they often coexist in the same patient because they share the same main cause: cigarette smoking
- COPD is an obstructive lung disease, meaning there is increased resistance to airflow - the FEV1/FVC ratio drops below 0.7
2. Definition of Emphysema
Irreversible (permanent) enlargement of airspaces distal to the terminal bronchiole, accompanied by destruction of their walls, without obvious fibrosis.
- Damage is beyond the terminal bronchiole (in the acini/alveoli)
- The walls are destroyed (not just stretched)
- There is no fibrosis - this distinguishes it from other lung conditions
3. Types of Emphysema
| Type | Location Affected | Key Association |
|---|---|---|
| Centriacinar (Centrilobular) | Proximal/central acini (respiratory bronchioles) - distal alveoli spared | Heavy smokers, upper lobes; >95% of cases |
| Panacinar (Panlobular) | Entire acinus uniformly - from respiratory bronchiole to terminal alveoli | α1-antitrypsin (α1-AT) deficiency; lower lobes |
| Paraseptal (Distal Acinar) | Distal part of acinus - near pleura and septa | Spontaneous pneumothorax in young adults; forms bullae |
| Irregular | Irregularly distributed - always with scarring/fibrosis | Clinically insignificant in most cases |
4. Pathogenesis
Main Causes
- Tobacco smoking (accounts for ~80% of COPD)
- α1-antitrypsin (α1-AT) deficiency (genetic cause)
Four Mechanisms
A. Toxic Injury and Inflammation
- Cigarette smoke damages respiratory epithelium
- Resident macrophages and epithelial cells release inflammatory mediators: leukotriene B4, IL-8, TNF
- These attract neutrophils from the circulation
- Neutrophils amplify inflammation and cause structural damage via growth factors and pro-inflammatory cytokines
B. Protease-Antiprotease Imbalance (Most Important)
- Nicotine acts as a chemoattractant for neutrophils
- Neutrophils release elastase (a protease) and reactive oxygen species (ROS)
- Elastase breaks down elastin - the elastic tissue that normally holds small airways open via radial traction during expiration
- Normally, α1-antitrypsin (produced by the liver) inhibits elastase
- In smokers or those with α1-AT deficiency: elastase exceeds inhibitor → elastic tissue destruction → airways collapse on expiration → airflow obstruction
C. Oxidative Stress
- Tobacco smoke and inflammatory cells produce oxidants (ROS) causing further tissue damage
- NRF2 is a transcription factor that normally activates antioxidant defense genes (including glutathione)
- Genetic variation in NRF2 is linked to increased susceptibility to smoking-related lung disease
D. Infection
- Does NOT initiate tissue destruction
- Superimposed bacterial/viral infections worsen existing inflammation and chronic bronchitis
5. Why Does Airflow Get Obstructed?
- Loss of elastic tissue → loss of radial traction → small airways collapse during expiration
- Goblet cell hyperplasia → excess mucus plugging airways
- Inflammatory infiltrates in bronchial walls (neutrophils, macrophages, B cells, T cells)
- Smooth muscle hypertrophy + peribronchial fibrosis → thickening of bronchiolar walls
6. α1-Antitrypsin Deficiency (Genetic Emphysema)
- Autosomal recessive disorder
- Encoded at the Pi locus on chromosome 14q
- Pi ZZ genotype (homozygous Z allele): serum α1-AT drops to <15% of normal
- ~1% of all emphysema patients have this defect
-
80% of Pi ZZ individuals develop panacinar emphysema - earlier in life and more severe if they smoke
- α1-AT is produced by hepatocytes, bone marrow cells, and lymphoid cells
- Also causes liver disease (protein accumulates in hepatocytes)
7. Morphology (What You See)
Gross (Naked Eye)
- Voluminous, enlarged lungs - often overlap and hide the heart
- Upper two-thirds more severely affected
- Blebs (<1 cm) and bullae (>1 cm) - large air-filled cysts, especially at the apex
Microscopic (Under the Microscope)
- Abnormally large alveoli separated by thin, destroyed septa
- Loss of alveolar attachments to small airway walls
- Pores of Kohn (inter-alveolar openings) become so large that septa appear to be "floating" in airspace
- Inflammatory changes around small airways
8. Clinical Features (Signs & Symptoms)
| Feature | Explanation |
|---|---|
| Dyspnea | Insidious onset, progressive - usually first symptom |
| Barrel chest | Hyperinflated lungs push ribs outward |
| Prolonged expiration | Patient hunches forward, breathes through pursed lips |
| Wheezing | Narrowed, collapsing airways |
| Cough | Due to mucus and inflammation |
| "Pink Puffer" | Over-ventilates to maintain near-normal blood gases; pink (not cyanosed), breathless |
- Narrowed airways → can't fully exhale → residual air builds up → hyperinflation
9. Complications
- Pulmonary arterial hypertension - from chronic hypoxia causing prolonged vasoconstriction
- Cor pulmonale (right heart enlargement from pulmonary hypertension) → right-sided heart failure
- Pneumothorax - especially from rupture of bullae (particularly in paraseptal emphysema in young adults)
- Coronary artery disease
- Respiratory failure
- Right-sided heart failure
- Massive lung collapse (pneumothorax)
10. Diagnosis
| Test | Finding |
|---|---|
| Arterial blood gas | Low O₂, high CO₂ |
| Chest X-ray | Hyperinflation, flattened diaphragm, bullae |
| Spirometry (gold standard) | Reduced FEV1, FEV1/FVC <0.7 - confirms airflow obstruction |
11. Treatment
- Smoking cessation - most important intervention
- Oxygen therapy
- Long-acting bronchodilators + inhaled corticosteroids
- Chest physiotherapy
- Surgical options:
- Bullectomy (removal of bullae)
- Lung volume reduction surgery
- Lung transplantation
- α1-AT replacement therapy - under evaluation for genetic cases
12. Other Special Forms
| Form | Description |
|---|---|
| Compensatory hyperinflation | Remaining alveoli expand to fill space after surgical removal of lung/lobe (not true emphysema) |
| Obstructive overinflation | Air trapped by partial airway blockage (tumor, foreign body) - can be life-threatening |
| Bullous emphysema | Large bullae (>1 cm) near apex; can rupture → pneumothorax |
| Interstitial emphysema | Air enters connective tissue stroma, mediastinum, or subcutaneous tissue (surgical emphysema) |
Quick Summary
Emphysema = permanent destruction of alveolar walls distal to terminal bronchioles → air trapping → hyperinflation → obstructive airflow pattern. Main causes are smoking (centriacinar type) and α1-AT deficiency (panacinar type). The core mechanism is elastase-antiprotease imbalance destroying lung elastic tissue. Complications include cor pulmonale, respiratory failure, and pneumothorax.
Make a visual diagram showing the protease-antiprotease imbalance mechanism
Medical educational diagram showing the protease-antiprotease imbalance mechanism in emphysema. The diagram should be a clean flowchart/pathway diagram with two parallel columns or pathways: LEFT SIDE - Normal (Balanced) State: - Cigarette smoke / noxious particles - Arrow down to: Neutrophils recruited to lung - Neutrophils release: Elastase (protease) - Balanced scale icon showing: Elastase ←→ α1-Antitrypsin - Result: Elastin intact, small airways held open, normal expiration RIGHT SIDE - Imbalanced State (Emphysema): - Cigarette smoke (heavy) OR α1-Antitrypsin deficiency (genetic, chromosome 14q, Pi ZZ genotype) - Arrow down to: Excess neutrophil recruitment + ROS production - Excess Elastase released, insufficient α1-Antitrypsin - Tipped/broken scale showing Elastase >> α1-AT - Elastin destroyed in alveolar walls - Loss of radial traction on small airways - Airways collapse during expiration - Air trapping → Hyperinflation → Permanent alveolar wall destruction = EMPHYSEMA Color coding: Green for normal/protective elements, Red/orange for damaging elements, Blue for structural lung tissue. Clear arrows showing progression. Title at top: "Protease-Antiprotease Imbalance in Emphysema". Clean white background, medical textbook style.
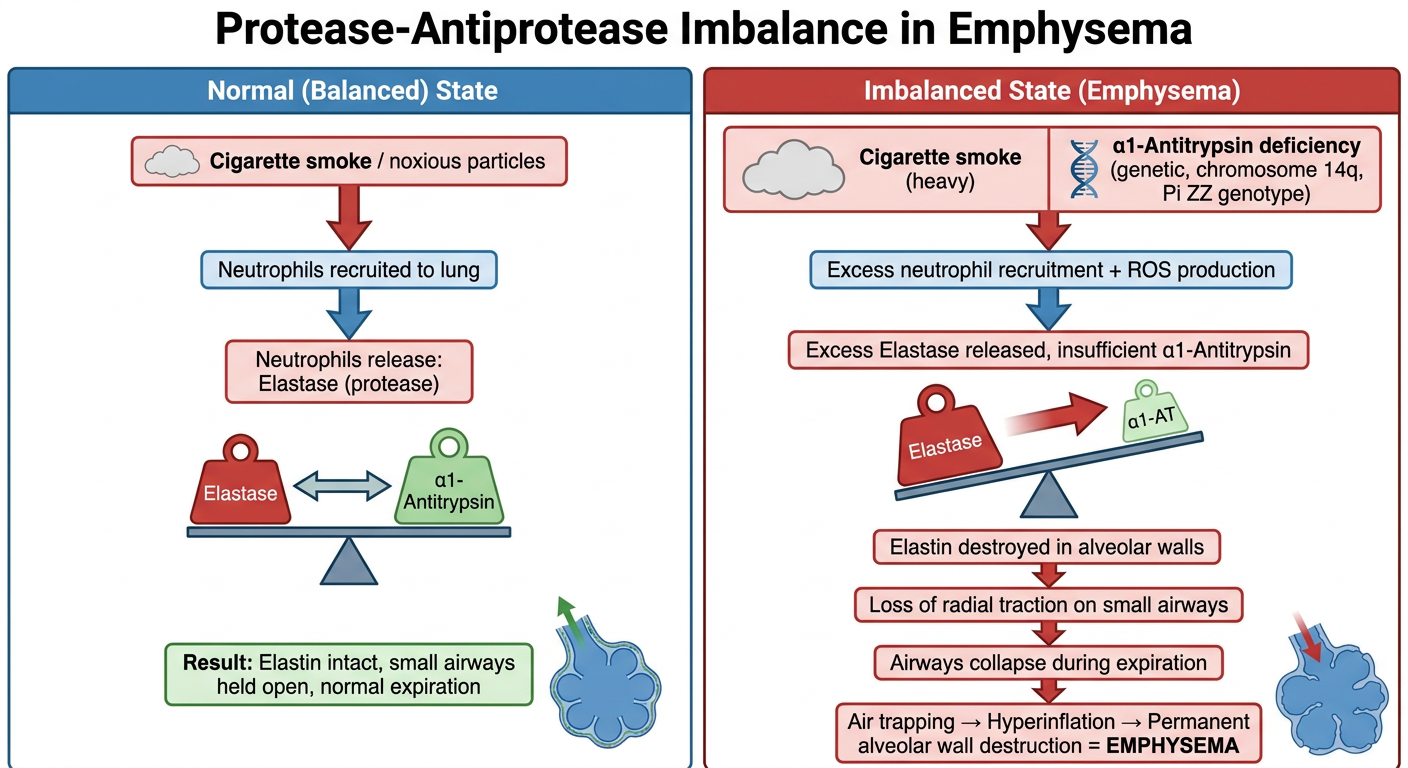
- Cigarette smoke recruits neutrophils, which release elastase
- α1-Antitrypsin (produced by the liver) keeps elastase in check
- Elastin is preserved, small airways stay open via radial traction, and expiration is normal
- Heavy smoking OR genetic α1-AT deficiency (Pi ZZ genotype, chromosome 14q) tips the balance
- Elastase overwhelms the available α1-AT
- Elastin in alveolar walls is digested and destroyed
- Without elastic support, small airways collapse during expiration
- Air traps in the lungs → hyperinflation → permanent alveolar wall destruction = Emphysema
what causes alveolar wall destruction
1. Protease-Antiprotease Imbalance (Primary Mechanism)
- Cigarette smoke and noxious particles recruit neutrophils and macrophages into the lung
- These cells release proteases - mainly elastase and matrix metalloproteinases (MMPs)
- Elastase directly digests elastin, the structural protein that makes up the alveolar wall framework
- Normally, α1-antitrypsin neutralizes elastase - but in smokers or Pi ZZ individuals, elastase activity overwhelms the inhibitor
- Result: the connective tissue scaffold of alveolar walls is broken down and cannot be repaired
2. Oxidative Stress
- Tobacco smoke and activated inflammatory cells (neutrophils, macrophages) flood the lung with reactive oxygen species (ROS)
- ROS directly damage alveolar epithelial cells and endothelial cells
- ROS also inactivate α1-antitrypsin, making the protease-antiprotease imbalance even worse
- The antioxidant defense (controlled by NRF2) is overwhelmed in heavy smokers
3. Alveolar Cell Apoptosis (Programmed Cell Death)
- Oxidative stress and inflammatory mediators trigger apoptosis (cell death) of:
- Type I pneumocytes (cover alveolar surface)
- Type II pneumocytes (repair and surfactant-producing cells)
- Capillary endothelial cells
- When these cells die faster than they can be replaced, the alveolar wall physically disintegrates
- This is a key reason why emphysematous damage is irreversible - the repair machinery is itself destroyed
4. Chronic Inflammation
- Persistent inflammatory cells in the alveolar walls release:
- TNF-α, IL-8, leukotriene B4 - sustain neutrophil/macrophage recruitment
- Growth factors that paradoxically cause structural remodeling rather than proper repair
- This chronic low-grade inflammation keeps the destructive cycle going even after smoking stops
The End Result
| What Gets Destroyed | Consequence |
|---|---|
| Elastin fibers | Loss of elastic recoil and radial traction |
| Alveolar septa | Smaller air sacs merge into large spaces |
| Capillary bed | Reduced gas exchange surface area |
| Airway support structures | Airways collapse on expiration |
gross and microscopy
Gross Morphology (Naked Eye)
Overall Appearance
- Lungs are voluminous and over-inflated - they are larger and heavier than normal
- They overlap and hide the heart when the chest is opened (a classic finding)
- Lungs feel spongy and may not collapse normally when the chest is opened
Distribution
- Upper two-thirds of the lungs are more severely affected (especially in centriacinar/smoking-related emphysema)
- Panacinar (α1-AT deficiency) preferentially affects the lower lobes and bases
Surface Features
- Blebs - air-filled spaces less than 1 cm, just under the pleura (subpleural)
- Bullae - larger air-filled cysts greater than 1 cm, often at the apex
- Bullae are especially characteristic of:
- Paraseptal (distal acinar) emphysema
- Irregular emphysema secondary to scarring
Cut Surface
- Large, abnormally wide alveoli visible to the naked eye
- Thin, wispy alveolar walls between the enlarged spaces
- Loss of the normal fine sponge-like texture
Microscopic Morphology (Histology)
Alveolar Changes
- Abnormally enlarged alveoli - the defining feature
- Thinned and destroyed alveolar septa - walls are sparse and fragile
- Loss of alveolar attachments to the outer wall of small airways - the airways lose their structural support
Pores of Kohn
- The inter-alveolar pores (Pores of Kohn) become so enlarged that adjacent alveoli merge
- Septa appear to be "floating" freely in airspace, or protrude blindly into the lumen with a club-shaped end - a hallmark microscopic sign
Vascular Changes
- Reduced capillary bed in alveolar walls
- Thickening of pulmonary arterial walls in advanced disease (due to pulmonary hypertension)
Airway / Inflammatory Changes
- Inflammatory infiltrates in bronchial and bronchiolar walls:
- Neutrophils, macrophages, B cells (sometimes forming follicles), T cells
- Goblet cell hyperplasia - excess mucus-secreting cells
- Smooth muscle hypertrophy in bronchiolar walls
- Peribronchial fibrosis - thickening of tissue around small airways
Side-by-Side Comparison
| Feature | Normal Lung | Emphysematous Lung |
|---|---|---|
| Alveolar size | Small, uniform | Large, irregular, merged |
| Alveolar wall | Intact, thin but continuous | Thinned, fragmented, destroyed |
| Septa | Connect alveoli firmly | Floating, club-ended remnants |
| Capillaries | Rich network in walls | Reduced, sparse |
| Airways | Open, supported | Collapsed, inflamed, thickened walls |
| Lung volume | Normal | Dramatically increased |
| Appearance | Fine sponge texture | Coarse, large holes visible |
Key Exam Points
- Gross: Voluminous lungs, hidden heart, subpleural bullae at apex
- Micro: Enlarged alveoli + thin destroyed septa + floating/club-ended septal remnants + inflammatory infiltrates in airways
- The absence of fibrosis microscopically is what separates emphysema from other destructive lung conditions
- Bullae >1 cm rupturing into the pleural space = spontaneous pneumothorax (classic in young adults with paraseptal emphysema)
why do blebs and bullae form
Step-by-Step Formation
Step 1 - Alveolar Wall Destruction
- Elastase and ROS destroy the walls between adjacent alveoli
- Multiple small alveoli lose their separating septa and merge into one larger space
- This is called confluence - the merged space holds more air than any single alveolus
Step 2 - Ball-Valve / Check-Valve Effect
- The small airways feeding these enlarged spaces become partially obstructed (inflamed walls, mucus, loss of elastic support)
- Air can flow in during inspiration (airways are pulled open by negative pressure)
- But during expiration, the already-collapsing airway closes before all the air escapes
- Result: more air enters than leaves with each breath cycle - progressive air trapping
Step 3 - Progressive Pressure Build-Up
- With each breath, a little more air accumulates in the trapped space
- The pressure inside the merged airspace gradually exceeds the surrounding lung pressure
- The thin, weakened walls of the space stretch outward
Step 4 - Bleb or Bulla Forms
- The expanding air pocket pushes outward toward the pleural surface (path of least resistance)
- It dissects through the loose connective tissue just under the visceral pleura
- A bleb forms when this air pocket is small (<1 cm) - essentially a subpleural air bubble
- With continued air trapping and expansion, the bleb grows into a bulla (>1 cm)
Why the Subpleural Location?
- Central lung parenchyma is surrounded and reinforced by adjacent alveoli on all sides
- Peripheral alveoli near the pleura have no support on one side
- Air preferentially dissects into the subpleural plane
- This is why bullae are almost always found at the apex and along pleural surfaces
Why the Apex Specifically?
- Pleural pressure is most negative (most sub-atmospheric) at the apex due to gravity
- This means apical alveoli are already subjected to greater distending pressure at baseline
- Any air trapping amplifies this further at the apex
- Blood flow is also lowest at the apex, so inflammatory repair is poorest there
Blebs vs Bullae - The Distinction
| Feature | Bleb | Bulla |
|---|---|---|
| Size | <1 cm | >1 cm |
| Wall | Visceral pleura only | Has remnants of lung tissue in wall |
| Contents | Air only | Air only |
| Origin | Subpleural dissection | Confluent destroyed alveoli |
| Main risk | Pneumothorax | Pneumothorax + compress surrounding lung |
Which Type of Emphysema Forms Bullae?
| Type | Bulla Formation |
|---|---|
| Paraseptal (distal acinar) | Most characteristic - multiple continuous enlarged spaces along septa and pleura; classic cause of spontaneous pneumothorax in young adults |
| Irregular emphysema | Bullae form adjacent to areas of fibrosis/scarring |
| Centriacinar (severe) | Can form large apical bullae in advanced disease |
| Panacinar | Less common, but can occur at lung bases |
The Danger - Pneumothorax
- Air escapes into the pleural space (which is normally a potential space under negative pressure)
- The negative pressure of the pleural space is lost
- The lung collapses on that side
- This is spontaneous pneumothorax - the classic complication of bullous/paraseptal emphysema, typically in tall, thin young men with apical bullae