bleeding disorders ? test for diagnosis essay
Reading File
Finding Sources
Finding Sources
Searching PubMed
"bleeding disorders" AND "diagnosis" AND "laboratory tests"
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
I now have comprehensive material to write a thorough essay. Let me compile it.
Bleeding Disorders: Classification and Diagnostic Tests
Introduction
Bleeding disorders, also termed hemorrhagic diatheses, are conditions characterized by abnormal bleeding that may appear spontaneously or follow an inciting event such as trauma or surgery. They arise from abnormalities in one or more components of the hemostatic system: the vessel wall (and its supportive connective tissue), platelets, or coagulation factors — alone or in combination. A systematic approach to diagnosis requires both clinical assessment and targeted laboratory investigations.
Classification of Bleeding Disorders
Bleeding disorders are broadly classified into four mechanistic categories:
1. Vessel Wall Abnormalities
Defects in the vessel wall or its surrounding connective tissue cause increased vascular fragility. Classic presentations include spontaneous petechiae and ecchymoses in the skin and mucous membranes, typically without abnormal coagulation tests. Causes include:
- Vitamin C deficiency (scurvy) — collagen defects weaken vessel walls
- Amyloidosis — amyloid deposition in blood vessel walls
- Ehlers-Danlos syndrome — inherited connective tissue disorder
- Chronic glucocorticoid use — acquired vascular fragility, particularly in older adults ("senile purpura")
- Infections — meningococcemia, rickettsioses, and septicemia cause microvascular damage or vasculitis
- Drug-induced hypersensitivity vasculitis — immune complex deposition in vessel walls
- Henoch-Schönlein purpura — IgA-mediated leukocytoclastic vasculitis
Key diagnostic clue: Platelet count and coagulation tests (PT, PTT) are typically normal; diagnosis is based on clinical findings and skin biopsy where appropriate.
2. Platelet Disorders
A. Thrombocytopenia (Quantitative Defects)
A platelet count below 150,000/µL is thrombocytopenia. Clinical bleeding risk rises significantly when counts fall to 20,000–50,000/µL, and spontaneous bleeding (especially intracranial) becomes a major hazard below 5,000/µL. The presentation is dominated by petechiae, purpura, and mucosal bleeding.
Causes are classified as:
| Mechanism | Examples |
|---|---|
| Decreased production | Aplastic anemia, marrow infiltration (leukemia), megaloblastic anemia, alcohol, cytotoxic drugs, HIV |
| Increased immunologic destruction | ITP, SLE, drug-induced (heparin, quinidine, sulfa), infectious mononucleosis |
| Increased non-immunologic destruction | DIC, TTP, HUS, microangiopathic hemolytic anemia |
| Sequestration | Hypersplenism |
| Dilutional | Massive transfusion |
Immune Thrombocytopenic Purpura (ITP): Caused by autoantibodies (usually IgG) against platelet membrane glycoproteins (GpIIb/IIIa or GpIb/IX). The spleen is the dominant site of platelet destruction. Diagnosis requires excluding secondary causes; bone marrow biopsy shows increased megakaryocytes.
Thrombotic Thrombocytopenic Purpura (TTP): Caused by deficiency of ADAMTS13, a plasma metalloprotease that cleaves ultra-large vWF multimers. Without cleavage, these multimers cause spontaneous platelet aggregation and microvascular thrombosis. Classic pentad: thrombocytopenia, microangiopathic hemolytic anemia (MAHA), neurological symptoms, renal failure, fever.
Hemolytic Uremic Syndrome (HUS): Similar to TTP but ADAMTS13 is normal. Typical HUS follows E. coli O157:H7 infection (Shiga-like toxin); atypical HUS involves complement regulatory protein mutations. Predominantly affects children and presents with bloody diarrhea → MAHA + renal failure.
B. Qualitative Platelet Defects
These disorders feature a normal platelet count but abnormal platelet function, often presenting with mucocutaneous bleeding patterns similar to thrombocytopenia.
- Bernard-Soulier syndrome — deficiency of GpIb/IX (platelet adhesion receptor for vWF)
- Glanzmann thrombasthenia — deficiency of GpIIb/IIIa (fibrinogen receptor for aggregation)
- Uremia — acquired platelet dysfunction (guanidinosuccinic acid accumulates and impairs platelet activation)
- Aspirin/NSAIDs — irreversible/reversible inhibition of COX → impaired thromboxane A₂ synthesis
3. Coagulation Factor Disorders
Defects in clotting factors prolong the PT, PTT, or both. Unlike platelet disorders, petechiae are typically absent. Instead, hemorrhage occurs in tissues subject to mechanical stress — particularly hemarthroses (joint bleeds), deep muscle hematomas, and massive post-surgical bleeding.
Von Willebrand Disease (vWD)
The most common inherited bleeding disorder (~1% of the population). Transmitted as autosomal dominant. Compound defect: reduced platelet adhesion (vWF links platelets to subendothelial collagen via GpIb) AND reduced factor VIII stabilization. Most patients present with mucocutaneous bleeding and menorrhagia.
Subtypes:
- Type I (most common): Quantitative reduction of vWF; autosomal dominant
- Type II: Selective loss of high-molecular-weight multimers (most active form); types IIA and IIB
- Type III: Severe quantitative deficiency; autosomal recessive; severe bleeding resembling hemophilia
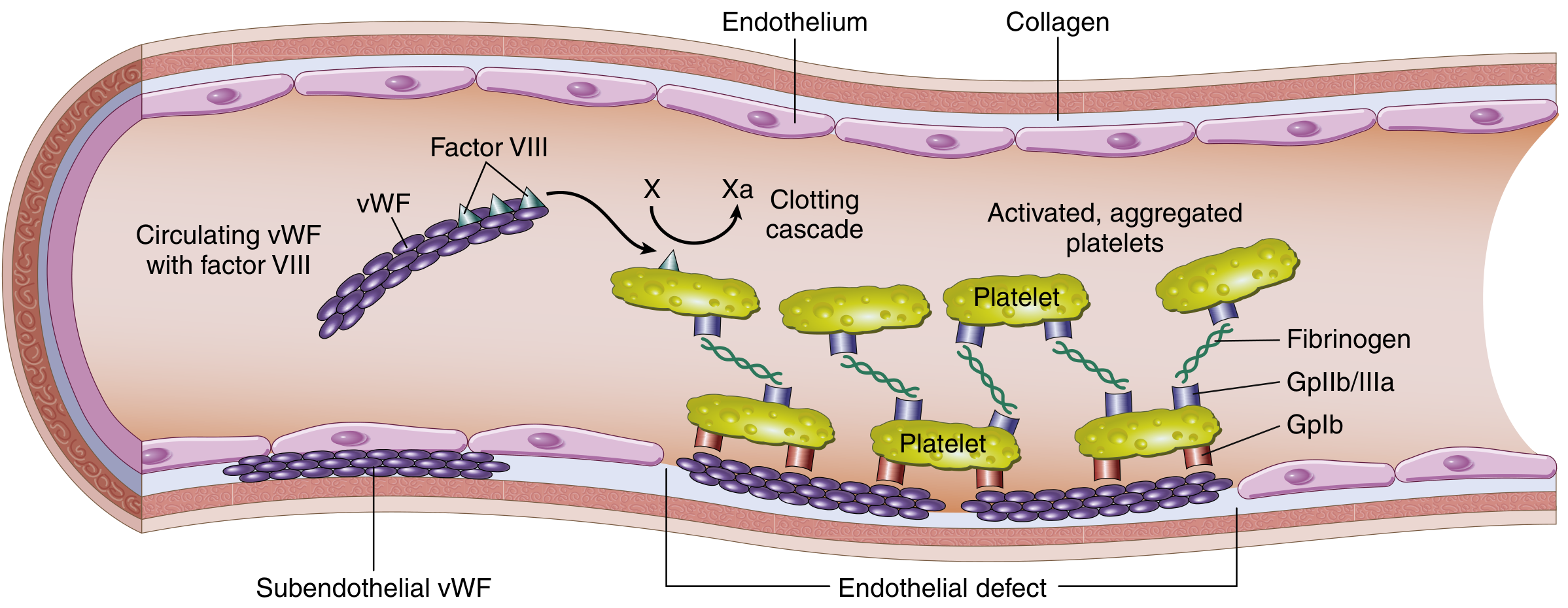
Hemophilia A (Factor VIII Deficiency)
The most common hereditary cause of serious bleeding. X-linked recessive; affects males primarily. Severity correlates with residual factor VIII activity:
- Severe: <1% activity → spontaneous hemarthroses, deep muscle bleeds
- Moderate: 1–5% activity → bleeding with minor trauma
- Mild: 5–30% activity → bleeding only with significant trauma or surgery
Typical findings: prolonged PTT, normal PT. Specific factor VIII assays confirm diagnosis. The PTT is corrected by mixing patient plasma with normal plasma (mixing study), unless inhibitors (neutralizing IgG antibodies) are present — seen in ~15% of severe cases.
Hemophilia B (Factor IX Deficiency — Christmas Disease)
Clinically indistinguishable from hemophilia A; also X-linked recessive. Much less common (~1:5 ratio vs. Hemophilia A). PTT prolonged, PT normal. Confirmed by specific factor IX assays.
Hemophilia C (Factor XI Deficiency)
Autosomal recessive; more prevalent in Ashkenazi Jews. Bleeding is generally mild and correlates poorly with factor level.
4. Combined / Acquired Disorders
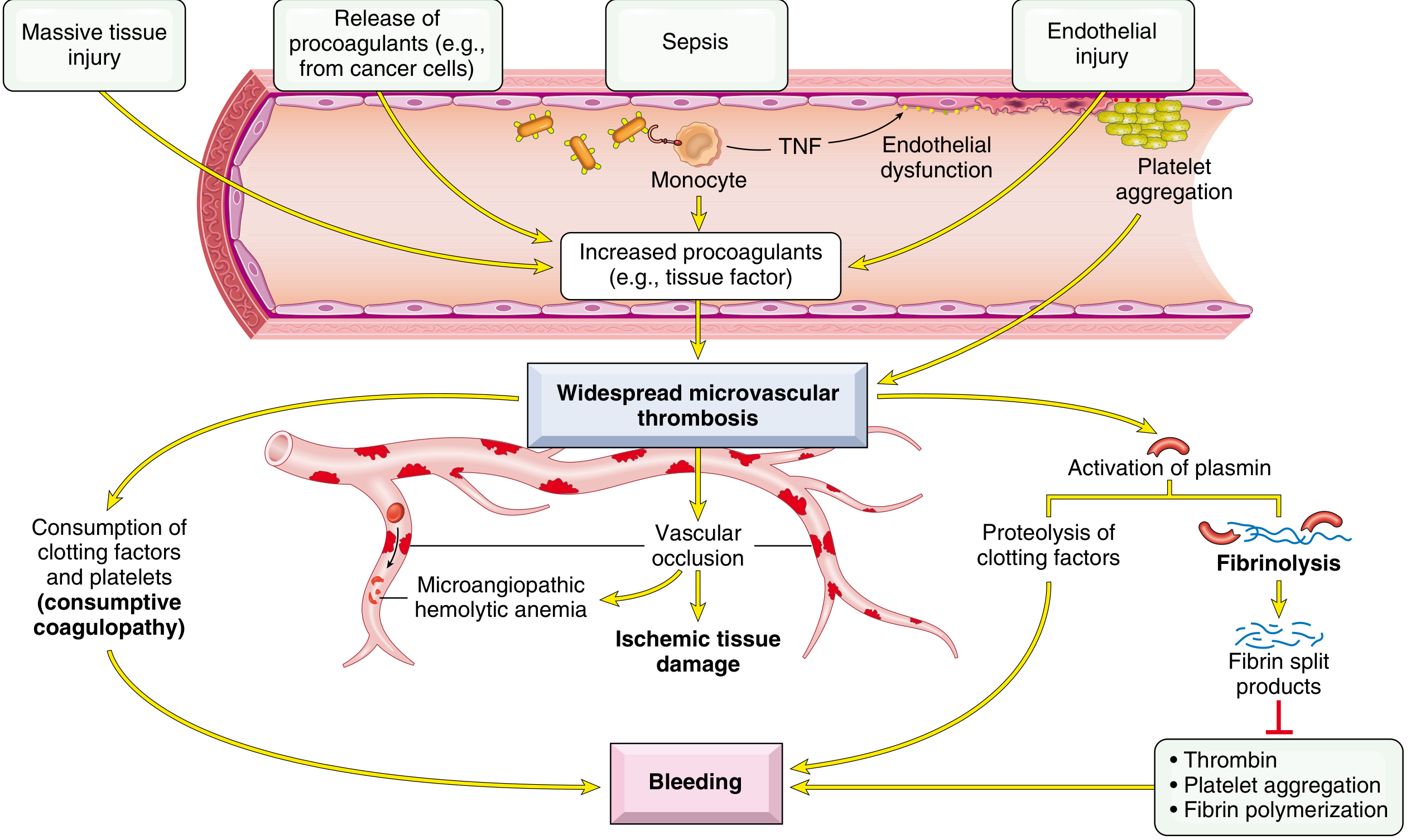
Disseminated Intravascular Coagulation (DIC)
DIC is a consumptive coagulopathy in which systemic coagulation activation simultaneously leads to:
- Microvascular thrombosis → ischemia of brain, kidneys, adrenals, lungs
- Consumption of platelets and clotting factors + fibrinolysis activation → severe hemorrhagic diathesis
Triggers include:
- Sepsis (endotoxin stimulates tissue factor expression on monocytes/endothelium)
- Major trauma, burns, extensive surgery
- Obstetric complications (amniotic fluid embolism, abruptio placentae, retained dead fetus)
- Malignancy (especially acute promyelocytic leukemia, pancreatic/lung adenocarcinomas)
- Antigen-antibody complexes (SLE, transfusion reactions)
Pathogenesis: Tissue factor or endothelial damage → thrombin generation throughout microcirculation → fibrin thrombi → ischemic microinfarcts + consumption of fibrinogen, factors V and VIII, and platelets. Plasmin activation causes fibrinolysis → fibrin degradation products (FDPs) including D-dimers, which further inhibit platelet aggregation and thrombin function.
Liver Disease
The liver synthesizes most clotting factors. Severe hepatic failure causes reduced synthesis of factors II, V, VII, IX, X, and fibrinogen → prolonged PT and PTT. Concurrent hypersplenism may also cause thrombocytopenia.
Vitamin K Deficiency
Vitamin K is required for γ-carboxylation (activation) of factors II, VII, IX, X and proteins C and S. Deficiency prolongs PT (factor VII has shortest half-life, affected first), then PTT. Causes: malabsorption, antibiotic use (gut flora reduction), neonatal hemorrhagic disease, warfarin overdose.
Laboratory Tests for Diagnosis
1. Prothrombin Time (PT) / INR
- What it tests: Extrinsic and common pathways (factors VII, X, V, II, fibrinogen)
- Method: Plasma + tissue thromboplastin (brain extract) + Ca²⁺ → clotting time in seconds
- Prolonged in: Factor VII, X, V, II, or fibrinogen deficiency; vitamin K deficiency; liver disease; warfarin; early DIC; lupus anticoagulant (sometimes)
- Expressed as INR (International Normalized Ratio) for warfarin monitoring standardization
- Normal PT: ~11–15 seconds
2. Partial Thromboplastin Time (PTT / aPTT)
- What it tests: Intrinsic and common pathways (factors XII, XI, IX, VIII, X, V, II, fibrinogen)
- Method: Plasma + kaolin (contact activator of factor XII) + cephalin (platelet phospholipid substitute) + Ca²⁺ → clotting time in seconds
- Prolonged in: Hemophilia A (factor VIII↓), hemophilia B (factor IX↓), hemophilia C (factor XI↓), factor XII deficiency (no bleeding), heparin therapy, DIC, lupus anticoagulant
- Normal aPTT: ~25–35 seconds
| Disorder | PT | PTT |
|---|---|---|
| Hemophilia A or B | Normal | Prolonged |
| Vitamin K deficiency (early) | Prolonged | Normal |
| Vitamin K deficiency (late) | Prolonged | Prolonged |
| Warfarin therapy | Prolonged | Normal or mildly prolonged |
| Heparin therapy | Normal | Prolonged |
| DIC | Prolonged | Prolonged |
| Liver disease | Prolonged | Prolonged |
| vWD (most types) | Normal | Normal or slightly prolonged |
| Factor VII deficiency alone | Prolonged | Normal |
3. Platelet Count
- Method: Electronic particle counter on anticoagulated (EDTA) blood; must be confirmed by peripheral blood smear (PBSM) to exclude clumping artifact
- Reference range: 150,000–450,000/µL (some sources: 150–350 × 10³/µL)
- Thrombocytopenia: <150,000/µL
- Thrombocytosis: >450,000/µL (reactive vs. myeloproliferative)
4. Peripheral Blood Smear (PBS)
- Confirms platelet count accuracy
- Detects schistocytes (fragmented RBCs) in TTP/HUS/DIC — key finding in MAHA
- Large platelets suggest peripheral destruction (ITP, TTP)
- Giant platelets in Bernard-Soulier syndrome
5. Bleeding Time
- Historically used to assess platelet function and vascular integrity in vivo
- A small standardized incision is made in the forearm; time to cessation is measured
- Prolonged in: Thrombocytopenia, qualitative platelet defects (vWD, aspirin, uremia, Glanzmann thrombasthenia, Bernard-Soulier syndrome)
- Now largely abandoned — time-consuming, difficult to standardize, poor predictive value for surgical bleeding; replaced by PFA-100
6. Platelet Function Analyzer (PFA-100 / Closure Time)
- In vitro simulation of primary hemostasis; measures time for platelet plug to occlude a collagen/ADP or collagen/epinephrine membrane under high shear stress
- Prolonged closure time in vWD, aspirin use, platelet receptor defects
- More reproducible than bleeding time; useful screening test for platelet dysfunction
7. Platelet Aggregation Studies
- Platelets are exposed to agonists: ADP, collagen, thrombin, arachidonic acid, ristocetin
- Aggregation response measured by light transmittance (platelet-rich plasma turbidimetry) or impedance
- Ristocetin-induced platelet aggregation (RIPA):
- Absent in vWD type I/IIA (insufficient vWF)
- Enhanced at low ristocetin concentrations in vWD type IIB (hyperfunctional vWF)
- Absent in Bernard-Soulier syndrome (absent GpIb receptor)
- Absent aggregation to all agonists except ristocetin → Glanzmann thrombasthenia (GpIIb/IIIa deficiency)
8. von Willebrand Factor Tests
Multiple assays are needed to diagnose and classify vWD:
| Test | Information |
|---|---|
| vWF antigen (vWF:Ag) | Quantity of vWF protein (ELISA or immunoturbidimetry) |
| vWF ristocetin cofactor activity (vWF:RCo) | Functional activity — ability of vWF to support ristocetin-mediated platelet agglutination |
| vWF:Ag/RCo ratio | Distinguishes quantitative (type I) from qualitative (type II) defects |
| Factor VIII:C assay | Reduced in vWD (vWF stabilizes FVIII); markedly low in type III |
| vWF multimer analysis | Gel electrophoresis — identifies absence of high-molecular-weight multimers in type IIA/IIB |
9. Specific Clotting Factor Assays
- Chromogenic or one-stage clotting assays measure activity of individual factors
- Essential for confirming hemophilia: Factor VIII assay (hemophilia A), Factor IX assay (hemophilia B)
- Mixing study: Patient plasma mixed 1:1 with normal plasma:
- PTT corrects → factor deficiency (hemophilia)
- PTT does not correct → inhibitor present (factor VIII inhibitor, lupus anticoagulant)
10. D-Dimer and Fibrin Degradation Products (FDPs)
- Products of plasmin-mediated fibrin cleavage
- D-dimer: Specific for cross-linked fibrin degradation (elevated in DIC, DVT/PE, sepsis)
- FDPs (includes X, Y, D, E fragments): elevated in DIC and primary fibrinolysis
- D-dimers and FDPs are elevated in 80–90% of acute DIC cases
- Inhibit platelet aggregation, fibrin polymerization, and thrombin → perpetuate coagulopathy
11. Fibrinogen Level
- Synthesized in liver; consumed in DIC and primary fibrinolysis
- Low fibrinogen (<150 mg/dL) in DIC, liver failure, primary fibrinolysis, dysfibrinogenemia
- Elevated as an acute-phase reactant in many other conditions
12. Thrombin Time (TT)
- Measures conversion of fibrinogen → fibrin by adding exogenous thrombin to plasma
- Prolonged in: Hypofibrinogenemia, dysfibrinogenemia, heparin therapy, DIC (FDPs interfere), dabigatran
- Useful to distinguish fibrinogen defects from other coagulopathies
13. Bone Marrow Biopsy
- Evaluates megakaryocyte number and morphology
- Increased megakaryocytes → peripheral platelet destruction (ITP, TTP, hypersplenism)
- Decreased megakaryocytes → reduced production (aplastic anemia, marrow infiltration)
14. Flow Cytometry for Platelet Glycoproteins
- Directly quantifies GpIb (CD42b) and GpIIb/IIIa (CD41/CD61) on platelet surface
- Confirms Bernard-Soulier syndrome (absent CD42b) and Glanzmann thrombasthenia (absent CD41/CD61)
15. ADAMTS13 Activity and Inhibitor Assay
- Reduced ADAMTS13 activity (<10%) confirms TTP
- ADAMTS13 inhibitor (autoantibody) confirms acquired TTP; its absence suggests hereditary form
16. Genetic Testing
- Molecular analysis for specific mutations in factor VIII gene (hemophilia A — intron 22 inversion most common), factor IX gene, vWF gene
- Used for carrier detection and prenatal diagnosis
Diagnostic Algorithm Summary
Suspected bleeding disorder
↓
Clinical history: pattern of bleeding (mucocutaneous vs. deep),
onset (congenital vs. acquired), family history, drug history
↓
First-line tests: PT, PTT, Platelet count, PBS
↓
┌───────────────────────────────────────────────────┐
│ Isolated prolonged PTT, normal PT │
│ → Factor VIII, IX, XI assay + mixing study │
│ → vWF studies if mucocutaneous bleeding │
├───────────────────────────────────────────────────┤
│ Isolated prolonged PT, normal PTT │
│ → Factor VII deficiency, early vitamin K deficiency│
├───────────────────────────────────────────────────┤
│ Both PT and PTT prolonged │
│ → DIC (+ D-dimer, fibrinogen, FDPs) │
│ → Liver disease, severe vitamin K deficiency │
│ → Common pathway factor deficiency (V, X, II) │
├───────────────────────────────────────────────────┤
│ Low platelet count, normal PT/PTT │
│ → PBS for schistocytes │
│ → Bone marrow biopsy │
│ → ADAMTS13 if TTP suspected │
├───────────────────────────────────────────────────┤
│ Normal all tests, clinical bleeding present │
│ → PFA-100, platelet aggregation, vWF studies │
│ → Vessel wall disorder (clinical/biopsy) │
└───────────────────────────────────────────────────┘
Conclusion
Bleeding disorders span a spectrum from mild mucocutaneous purpura to life-threatening hemorrhage. Accurate diagnosis demands integration of the clinical bleeding pattern with targeted laboratory investigations. The PT and PTT define defects in the extrinsic and intrinsic pathways respectively; the platelet count and function tests evaluate primary hemostasis; and specialized assays — von Willebrand factor studies, specific factor assays, ADAMTS13, D-dimer, and genetic testing — confirm specific diagnoses. This systematic approach, moving from screening tests to confirmatory assays, is the cornerstone of modern management of hemorrhagic diatheses.
Sources:
- Robbins & Kumar Basic Pathology, 10th ed. (Elsevier) — Chapter 10: Bleeding Disorders, pp. 426–431
- Robbins, Cotran & Kumar Pathologic Basis of Disease, 10th ed. — Chapter 14: Bleeding Disorders, pp. 618–627
- Recent literature: Mehic et al. (2024), "Utility of Global Hemostatic Assays in Patients with Bleeding Disorders of Unknown Cause," Hämostaseologie [PMID: 38950624]; Gresele et al. (2025), "Investigation of Bleeding Disorders: When and How Should We Test Platelet Functions?" Hämostaseologie [PMID: 40355112]
This is a shared conversation. Sign in to Orris to start your own chat.