barter syndrome
Reading File
Finding Sources
Searching PubMed
"Bartter syndrome" diagnosis management
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information to give a thorough answer.
Bartter Syndrome
Bartter syndrome is a group of autosomal recessive renal tubular disorders defined by impaired salt reabsorption in the thick ascending limb (TAL) of the loop of Henle. The net result mimics chronic loop diuretic administration: hypokalemia, hypochloremic metabolic alkalosis, hyperreninemia, hyperaldosteronism, and normal-to-low blood pressure.
- Brenner and Rector's The Kidney, 2-Volume Set, p. 3151
- NKF Primer on Kidney Diseases, 8e, p. 176
Pathophysiology
The TAL normally reabsorbs ~25-30% of filtered NaCl without water, generating the medullary concentration gradient. Four key transport proteins accomplish this:
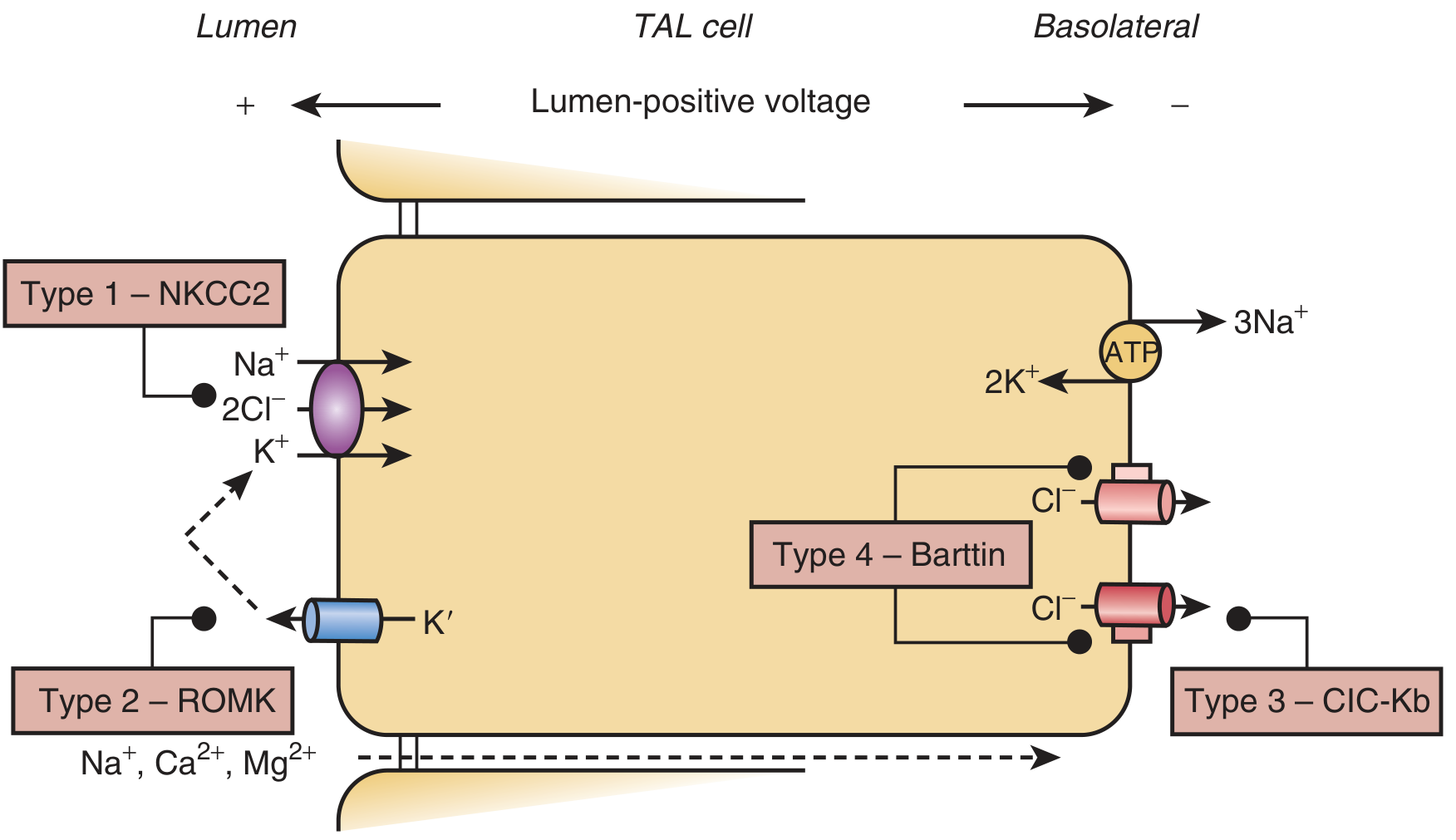
Loss of function in any of these proteins eliminates the lumen-positive transepithelial voltage, abolishes paracellular reabsorption of Na+, Ca2+, and Mg2+, and causes salt wasting. The resulting volume depletion activates the renin-angiotensin-aldosterone system (RAAS), delivering excess NaCl to the collecting tubule, where Na+ reabsorption drives K+ and H+ secretion - producing hypokalemia and metabolic alkalosis.
Additionally, defective chloride reabsorption at the macula densa impairs tubuloglomerular feedback (TGF) and activates COX-2, which upregulates prostaglandin E2 (PGE2), causing afferent arteriolar dilation, further hyperreninism, and polyuria.
Genetic Classification (Types 1-6)
| Type | Gene | Protein | Key Features |
|---|---|---|---|
| 1 | SLC12A1 | NKCC2 (apical Na-K-2Cl cotransporter) | Antenatal; polyhydramnios, nephrocalcinosis |
| 2 | KCNJ1 | ROMK (apical K+ channel) | Antenatal; paradoxical neonatal hyperkalemia (resolves in days-weeks) |
| 3 | CLCNKB | ClC-Kb (basolateral Cl- channel) | Classic postnatal; mildest; rarely nephrocalcinosis; can mimic Gitelman |
| 4a | BSND | Barttin (subunit for ClC-Ka + ClC-Kb) | Antenatal + sensorineural deafness; linked to chr 1p31 |
| 4b | CLCNKA + CLCNKB | Both ClC-Ka and ClC-Kb | Severe antenatal; deafness |
| 5 | CASR (gain-of-function) | Calcium-sensing receptor | Autosomal dominant hypocalcemia phenotype; CaSR inhibits ROMK |
| 6 | MAGED2 | MAGE-D2 (regulatory) | Transient antenatal form; resolves completely by 3-4 weeks of life; X-linked |
Types 1, 2, and 4 typically present antenatally (most severe); type 3 presents classically in childhood; types 5 and 6 are rarer variants.
Clinical Features
Antenatal / Neonatal Bartter
- Maternal polyhydramnios (fetal polyuria)
- Premature birth
- Postnatal: severe polyuria, vomiting, failure to thrive
- Hypercalciuria and nephrocalcinosis (especially types 1, 2, 4)
- Marked electrolyte wasting: hypokalemia, metabolic alkalosis
- Type 4: add sensorineural deafness
- Type 2 neonates: paradoxical hyperkalemia initially (ROMK also mediates K+ secretion in collecting duct; other channels compensate within the first week)
Classic (Postnatal) Bartter - predominantly Type 3
- Failure to thrive, polyuria, polydipsia
- Hypokalemic, hypochloremic metabolic alkalosis
- Normal to low blood pressure despite elevated renin/aldosterone
- Vascular resistance to angiotensin II
- ~20% hypomagnesemia
- Often hypercalciuria (but less than antenatal form)
- Additional findings: growth hormone deficiency, hypophosphatemia with renal phosphate wasting, hyperparathyroidism
Distinguishing Bartter from Gitelman Syndrome
| Feature | Bartter | Gitelman |
|---|---|---|
| Defective segment | TAL (loop of Henle) | DCT (distal convoluted tubule) |
| Affected protein | NKCC2, ROMK, ClC-Kb, Barttin | NCC (thiazide-sensitive cotransporter) |
| Mimics | Loop diuretics | Thiazide diuretics |
| Onset | Antenatal or childhood | Late childhood / adulthood |
| Calcium | Hypercalciuria, nephrocalcinosis | Hypocalciuria |
| Magnesium | Normal (or mild hypomagnesemia) | Hypomagnesemia (prominent) |
| Severity | More severe | Milder; salt craving, cramps, fatigue |
The urine chloride is elevated in Bartter syndrome (distinguishing it from surreptitious vomiting, where urine Cl- is low).
Diagnosis
- Biochemistry: hypokalemia, metabolic alkalosis, elevated plasma renin and aldosterone, normal-to-low blood pressure
- Urine: elevated Cl-, often hypercalciuria
- Exclude: surreptitious vomiting (low urine Cl-), diuretic abuse (urine diuretic screen), laxative abuse
- Genetic testing: confirms subtype
- Kidney ultrasound: may show nephrocalcinosis in antenatal types
Treatment
Treatment is supportive and does not correct the underlying tubular defect. Goals are to minimize hypokalemia and electrolyte losses.
- Salt and potassium supplementation - mainstay; IV saline in severely affected neonates
- Indomethacin (COX inhibitor) - reduces PGE2, improves polyuria, raises K+, decreases renin activity; especially useful in antenatal/neonatal forms. Risk of necrotizing enterocolitis and bowel perforation in neonates.
- Selective COX-2 inhibitors (rofecoxib, celecoxib) - proposed as alternatives with lower GI risk, though cardiovascular safety concerns exist with long-term use
- Potassium-sparing diuretics (amiloride, spironolactone, eplerenone) - help correct hypokalemia but worsen salt wasting; use with great caution, especially in infants
- ACE inhibitors / propranolol - reduce aldosterone-driven K+ losses
- Magnesium supplementation - when hypomagnesemia is present
Key Points
- Bartter syndrome = "loop diuretic in a gene" - loss of TAL salt reabsorption
- Always consider if a patient has persistent hypokalemic metabolic alkalosis + normal/low BP + elevated renin/aldosterone
- Type 4 = deafness (barttin mutation)
- Type 2 = paradoxical neonatal hyperkalemia
- Type 6 (MAGED2) = transient (resolves spontaneously)
- Prostaglandins amplify the phenotype - COX inhibition is a cornerstone of therapy in severe forms
- Gitelman vs. Bartter: calcium is the key discriminator (hypercalciuria vs. hypocalciuria)
Recent literature (2023 systematic review of case reports, PMID 37763757) continues to highlight phenotypic variability across subtypes and underscores the value of genetic confirmation.
explain this in much easier way
Bartter Syndrome - Simple Explanation
What is it?
Imagine your kidneys are like a recycling factory for salt. Normally, when blood is filtered, the kidneys reclaim most of the salt back into the body before it leaves in urine.
In Bartter syndrome, one of the recycling machines is broken. Salt leaks out into the urine instead of being saved. This one problem causes a chain reaction of issues throughout the body.
The Chain Reaction
Broken salt recycler in kidney
↓
Salt leaks into urine (salt wasting)
↓
Body thinks it's "low on volume/fluid"
↓
Hormones (renin, aldosterone) shoot up to compensate
↓
Aldosterone tells kidneys to dump K+ and H+ instead
↓
Low potassium (hypokalemia) + alkalosis
Simple Analogy
Think of the kidney tubule like a leaky bucket assembly line:
- Normally, workers (transport proteins) reclaim salt at every station
- In Bartter, the station in the middle (Loop of Henle) has a broken worker
- Salt floods downstream, overwhelms the later stations, and the body loses potassium trying to compensate
- Blood pressure stays low despite high alarm hormones, because the body never actually has enough salt
The 4 Broken Workers (Types 1-4)
All cause the same problem, just different proteins are broken:
| Type | Broken Part | Simple Name |
|---|---|---|
| 1 | NKCC2 | The main salt pump |
| 2 | ROMK | The potassium recycler |
| 3 | ClC-Kb | The chloride exit door |
| 4 | Barttin | The helper subunit for the exit doors - also causes deafness |
Who Gets It & When?
- Severe types (1, 2, 4): Problems start in the womb - baby urinates excessively, causing the amniotic fluid to overflow (polyhydramnios). Baby is born premature and very sick.
- Milder type (3): Problems show up in childhood - kid doesn't grow well, drinks too much water, muscles are weak.
What Does the Patient Look Like?
- Always tired and weak (low potassium affects muscles)
- Drinks a lot, pees a lot (can't concentrate urine)
- Poor growth in children
- Normal or low blood pressure - even though the body is producing huge amounts of blood pressure hormones (the body just doesn't respond to them properly)
- May have kidney stones/calcium deposits in severe types
- Type 4: also deaf
How is it Different from Gitelman Syndrome?
Both are "broken kidney recycler" diseases, but they break at different spots:
| Bartter | Gitelman | |
|---|---|---|
| Where broken | Loop of Henle (middle) | Distal tubule (near end) |
| Mimics | Loop diuretic (furosemide) | Thiazide diuretic |
| Calcium in urine | High (leaks out) | Low (saved too well) |
| Onset | Early (newborn/child) | Later (teen/adult) |
| Severity | More severe | Milder |
Easy memory trick: Bartter = Loop (of Henle) = Loop diuretic. Gitelman = distal = thiazide.
Treatment (Simple)
There's no cure - you manage the symptoms:
- Give extra salt and potassium - replace what's being lost
- Indomethacin (anti-inflammatory) - the kidneys in Bartter over-produce inflammatory signals (prostaglandins) that make things worse; blocking these helps reduce polyuria and potassium loss
- Potassium-sparing drugs (amiloride, spironolactone) - help hold on to potassium
- ACE inhibitors - dampen the overactive hormone system
One-Line Summary
Bartter syndrome is a genetic kidney leak where the salt recycler in the Loop of Henle is broken, causing the body to waste salt and potassium, triggering sky-high hormones - but blood pressure stays low because the leak is never fixed.
levels of scans and exmples in pregnancy
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
I now have enough comprehensive content. Here's the full easy explanation:
Levels of Scans in Pregnancy - Simple Guide
Think of pregnancy scans like progress checkpoints - each one checks something different depending on how far along the pregnancy is.
Overview - The Timeline
Week 6-10 Week 11-14 Week 18-22 Week 28-36+
| | | |
Early/Dating 1st Trimester Anomaly Scan Growth/
Scan Combined Scan (The Big Scan) Wellbeing Scan
Level 1 - Early / Dating Scan (6-10 Weeks)
What it checks:
- Is the pregnancy inside the uterus? (rules out ectopic)
- How many babies? (twins?)
- Is there a heartbeat?
- Measures the Crown-Rump Length (CRL) to confirm exact dates
Simple example:
"Patient comes in unsure of her dates. The scan shows a single fetus with CRL of 22mm = 9 weeks and 2 days exactly."
What it can find:
- Miscarriage (no heartbeat)
- Ectopic pregnancy (sac outside uterus = emergency)
- Twin/triplet pregnancy
- Blighted ovum (empty sac)
Level 2 - First Trimester Combined Scan (11-14 Weeks)
This is actually a scan + blood test together - that's why it's called "combined."
The scan part checks:
- Nuchal Translucency (NT) - measures a fluid pad at the back of the baby's neck
- A thicker NT = higher chance of Down syndrome or heart defects
- Normal NT = less than 3.5mm
The blood test part checks:
- PAPP-A (pregnancy-associated plasma protein A) - low in Down syndrome
- Free beta-hCG - high in Down syndrome
Combined, this detects:
- ~95% of Down syndrome (Trisomy 21) at a 5% false-positive rate
- Trisomy 18 (Edwards syndrome)
- Trisomy 13 (Patau syndrome)
Simple example:
"NT measured at 4.5mm (thick). Blood tests show low PAPP-A and high hCG. Combined risk = 1 in 50 for Down syndrome. Offered further testing (NIPT or amniocentesis)."
Level 3 - Anomaly Scan / "The Big Scan" (18-22 Weeks)
This is the most detailed structural scan. Every major organ is systematically checked.
What's measured (Biometry):
| Measurement | What it tells us |
|---|---|
| BPD (Biparietal Diameter) | Head width - confirms gestational age |
| HC (Head Circumference) | Overall head size |
| AC (Abdominal Circumference) | Baby's tummy size - monitors growth |
| FL (Femur Length) | Thigh bone length - checks proportions |
Organs checked one by one:
Head/Brain:
- Cerebral ventricles (fluid spaces - enlarged = hydrocephalus?)
- Cerebellum (banana-shaped = spina bifida sign)
- Cavum septi pellucidi (small space in midline brain)
- Cisterna magna
Face:
- Lips (cleft lip)
- Palate (cleft palate harder to see)
- Eyes, nose
Chest:
- 4-chamber heart view (size, position, chambers equal?)
- Outflow tracts (left and right ventricular outflow)
- Lungs (any masses?)
- Diaphragm intact?
Abdomen:
- Stomach bubble visible? (if absent = oesophageal atresia)
- Bowel loops (bright = cystic fibrosis marker?)
- Kidneys (both present, no hydronephrosis)
- Bladder visible?
- Abdominal wall - cord insertion normal? (exomphalos/gastroschisis?)
Spine:
- Full spinal column intact (open spina bifida = gap visible)
Limbs:
- All 4 limbs present
- Bone lengths normal
Placenta, Cord, Fluid:
- Placental position (low-lying = possible previa?)
- Cord = 3 vessels (2 arteries + 1 vein - only 2 vessels = kidney abnormality risk)
- Amniotic fluid volume
Simple examples:
"Scan at 20 weeks: stomach bubble absent. Diagnosis = likely oesophageal atresia (EA). Baby referred to tertiary centre."
"Scan at 20 weeks: posterior fossa shows 'banana sign' and 'lemon-shaped' head. Diagnosis = Arnold-Chiari malformation (associated with open spina bifida)."
"Placenta covers the cervix completely = placenta praevia. Mother warned about bleeding risk, planned C-section."
Level 4 - Growth / Wellbeing Scan (28 Weeks Onwards)
Done in high-risk pregnancies or when the anomaly scan raised concerns.
What it checks:
- Fetal growth - is the baby on its centile?
- Doppler flow studies:
- Umbilical artery (resistance = placenta struggling?)
- Middle cerebral artery (brain-sparing effect?)
- Ductus venosus (late sign of compromise)
- Amniotic fluid volume (too little = oligohydramnios; too much = polyhydramnios)
- Fetal movements and breathing movements (Biophysical Profile)
Simple example:
"Mother with hypertension at 32 weeks. Growth scan shows AC on 3rd centile (small). Umbilical artery Doppler shows absent end-diastolic flow = placental insufficiency. Decision made for early delivery."
Special/Advanced Level - Detailed/Tertiary Scan (Level 3 Ultrasound)
Done at specialist centres when something is found on routine scanning.
Extra things checked (on top of standard anomaly scan):
- Full cardiac survey (fetal echo)
- Brain: third & fourth ventricles, vermis, cisterna magna with measurements
- Nuchal fold thickness (15-20 weeks - Down syndrome marker)
- Ribs, palate in detail
- Looking for ascites
Simple example:
"Routine anomaly scan shows a bright bowel. Referred for detailed scan: confirmed echogenic bowel + dilated bowel loops. Concern for cystic fibrosis or Down syndrome. Offered amniocentesis."
Screening Blood Tests That Go With the Scans
| Test | When | What it screens |
|---|---|---|
| First trimester combined (NT + PAPP-A + hCG) | 11-14 weeks | Down syndrome (95% detection) |
| Quadruple screen (AFP + hCG + estriol + inhibin A) | 15-20 weeks | Down, Trisomy 18, neural tube defects |
| NIPT/cfDNA (cell-free fetal DNA in mum's blood) | From 10 weeks | Down, Trisomy 18, Trisomy 13, sex chromosomes (>99% accuracy) |
Quadruple screen pattern to remember:
| Condition | AFP | hCG | Estriol | Inhibin A |
|---|---|---|---|---|
| Down syndrome | ↓ Low | ↑ High | ↓ Low | ↑ High |
| Trisomy 18 | ↓ Low | ↓ Low | ↓ Low | Normal/↓ |
| Open neural tube defect | ↑↑ High | Normal | Normal | Normal |
Quick Summary - One Table
| Scan | Week | Main Purpose | Key Finding Example |
|---|---|---|---|
| Early / Dating | 6-10 wks | Confirm dates, heartbeat, location | Ectopic, twins, miscarriage |
| 1st Trimester Combined | 11-14 wks | Down syndrome screening | Thick NT = ↑ Down risk |
| Anomaly Scan | 18-22 wks | Check all organs/structure | Spina bifida, heart defect, cleft lip |
| Growth/Doppler | 28+ wks | Monitor size + placenta | Small baby, poor placental flow |
| Detailed/Tertiary | Any | Deep dive when problem found | Fetal echo, detailed brain |
- Creasy & Resnik's Maternal-Fetal Medicine, Principles and Practice
- Textbook of Family Medicine, 9e, p. 482
- Tietz Textbook of Laboratory Medicine, 7th ed., p. 2897
This is a shared conversation. Sign in to Orris to start your own chat.