Discuss in detail about the mechanism of enzyme induction and enzyme inhibition for 10 mark
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive, exam-ready answer on enzyme induction and enzyme inhibition, drawing from Goodman & Gilman's The Pharmacological Basis of Therapeutics, Lippincott Illustrated Reviews: Pharmacology, and Dermatology 2-Volume Set 5e.
Mechanism of Enzyme Induction and Enzyme Inhibition
Drug-metabolizing enzymes — chiefly the cytochrome P450 (CYP) superfamily and phase II conjugation enzymes — are highly susceptible to modification by drugs, dietary chemicals, and environmental xenobiotics. Two key phenomena arise: enzyme induction (increased enzyme activity → accelerated metabolism) and enzyme inhibition (reduced enzyme activity → slowed metabolism). Both carry major clinical consequences.
PART I — ENZYME INDUCTION
Definition
Enzyme induction is a gene-mediated increase in the synthesis or a slowing in the turnover of drug-metabolizing enzymes following chronic exposure to a xenobiotic (inducer). The net result is accelerated metabolism of the inducer itself (autoinduction) or of co-administered drugs.
— Goodman & Gilman's, p. 134
Mechanism
Step 1 — Binding to a Nuclear Receptor
Inducers do not act directly on the enzyme. They first bind to intracellular nuclear receptors, which act as ligand-activated transcription factors. The principal receptors are:
| Receptor | Key Ligand/Inducer |
|---|---|
| Aryl Hydrocarbon Receptor (AHR) | Omeprazole, polycyclic aromatic hydrocarbons |
| Pregnane X Receptor (PXR) | Rifampicin, dexamethasone, St. John's wort |
| Constitutive Androstane Receptor (CAR) | Phenobarbital |
| PPARα | Fibrates (gemfibrozil, fenofibrate) |
| Vitamin D Receptor (VDR) | Vitamin D |
| Retinoic Acid Receptor (RAR) | All-trans retinoic acid |
Step 2 — Receptor Activation and Heterodimerization
- For AHR: The ligand-bound AHR translocates to the nucleus, dimerizes with its nuclear partner (ARNT), and binds to xenobiotic response elements (XREs) upstream of target CYP genes (CYP1A1, CYP1A2, CYP1B1).
- For Type 2 nuclear receptors (PXR, CAR): After ligand binding, the receptor heterodimerizes with the Retinoid X Receptor (RXR), forming a PXR–RXR (or CAR–RXR) complex that binds to drug-response elements on the promoter region of target genes.
Step 3 — Recruitment of Coactivators and Transcription
The receptor–RXR complex recruits coactivator proteins (e.g., SRC-1, CBP/p300) → assembles the general transcription machinery (TBP, TATA box, RNA Pol II) → drives transcription of CYP genes (notably CYP3A4, CYP2B6, CYP2C9).
Step 4 — Increased Enzyme Synthesis
Enhanced mRNA → ribosomal translation → increased CYP protein in hepatic smooth ER → accelerated oxidative metabolism of substrate drugs.
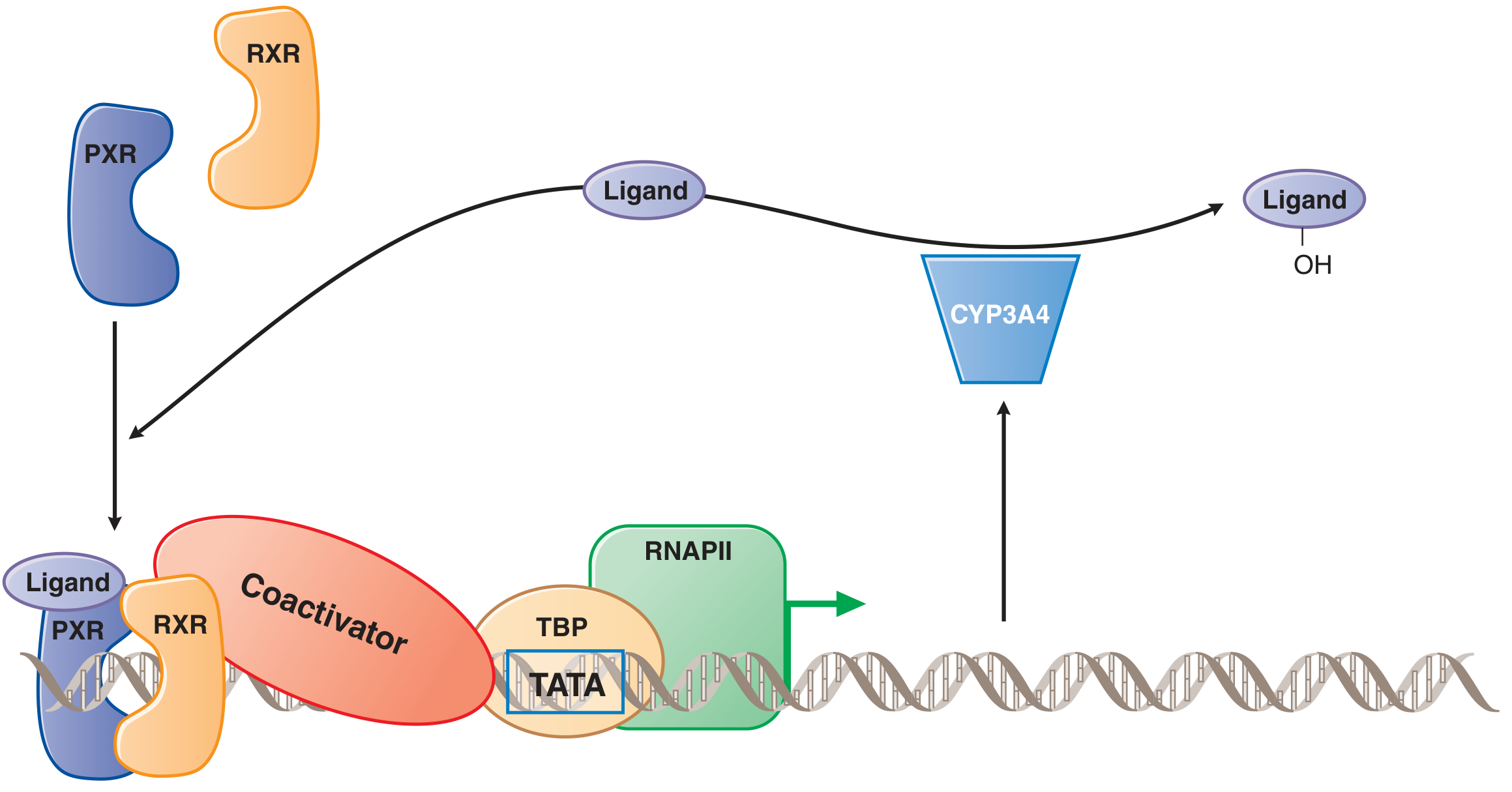
Figure: PXR–RXR mediated induction of CYP3A4. — Goodman & Gilman's, Fig. 5–13
Temporal Course
Induction is a slow process — onset over days to weeks of continued exposure. This is because it requires new protein synthesis. Similarly, the effect wanes gradually after stopping the inducer (enzyme levels return to baseline over days to weeks).
Clinical Consequences of Enzyme Induction
| Scenario | Example |
|---|---|
| Reduced efficacy of co-administered drug | Rifampicin induces CYP3A4 → accelerates metabolism of oral contraceptives → contraceptive failure |
| Loss of efficacy of the inducer itself (autoinduction) | Carbamazepine induces its own CYP3A4/CYP3A5-mediated metabolism |
| Increased toxicity via toxic metabolite production | Rifampicin + ritonavir: PXR-mediated induction of CYP3A4 → excess ritonavir toxic metabolites → hepatotoxicity |
| Activation of procarcinogens | Omeprazole induces CYP1A1/1A2 via AHR → activation of environmental procarcinogens |
| Drug–herbal interaction | St. John's wort (hyperforin) activates PXR → CYP3A4 induction → failure of oral contraceptives, tacrolimus, etc. |
Common Enzyme Inducers (CYP3A4)
"COPERS" — Carbamazepine, Oxcarbazepine, Phenytoin, Efavirenz, Rifampin, St. John's wort (also phenobarbital, dexamethasone, nevirapine)
— Lippincott Illustrated Reviews: Pharmacology, p. 60; Dermatology 5e, p. 131
PART II — ENZYME INHIBITION
Definition
Enzyme inhibition is the reduction or abolition of CYP-mediated drug metabolism by a drug or chemical, leading to increased plasma concentrations of co-administered substrate drugs, with risk of exaggerated response or toxicity.
"Inhibition of drug metabolism is the most important mechanism for drug interactions because it can lead to an increase in plasma drug concentration, enhanced drug response, and toxicity." — Dermatology 5e, p. 131
Temporal Course
Unlike induction, inhibition is rapid — it begins within the first one or two doses of the inhibitor and is maximal at steady-state concentrations of the inhibitor. The time course is therefore hours to days, not weeks.
Types of Enzyme Inhibition
1. Competitive (Reversible) Inhibition
- The inhibitor competes with the substrate for the same active site of the CYP enzyme.
- Drugs such as macrolide antibiotics (erythromycin, clarithromycin), cimetidine, and ketoconazole bind tightly to the heme iron of the CYP isoenzyme.
- As inhibitor concentration increases, the degree of isoenzyme saturation increases → at full saturation, the patient effectively behaves as a "poor metabolizer" of all substrates for that CYP.
- This is reversible — removing the inhibitor restores enzyme function.
Example: Ketoconazole inhibits CYP3A4 → reduced metabolism of cyclosporine → cyclosporine toxicity.
2. Non-competitive Inhibition
- The inhibitor binds at a different site from the substrate (allosteric site), causing a conformational change in the enzyme.
- The inhibitor can bind whether or not the substrate is present.
- Maximum reaction velocity (Vmax) is reduced; Km is unchanged.
- Some drugs inhibit CYP reactions for which they are not themselves substrates — e.g., ketoconazole inhibits multiple CYPs beyond the one that metabolizes it.
3. Mechanism-Based (Irreversible / Suicide) Inhibition
- Also called "suicide inhibition" or time-dependent inhibition.
- The inhibitor (or its reactive metabolite, generated by the CYP itself) covalently and irreversibly modifies the enzyme, permanently destroying its catalytic activity.
- Recovery requires de novo synthesis of new CYP enzyme (days).
- This results in destruction of the enzyme and release of heme, which is then catabolized by heme oxygenase → reduction in the free heme pool → secondary induction of δ-aminolevulinic acid synthase (ALAS1) — clinically relevant in porphyria triggers.
Examples:
- Erythromycin and clarithromycin form nitroso metabolites that covalently bind CYP3A4
- Grapefruit juice furanocoumarins irreversibly inhibit intestinal CYP3A4
4. Product Inhibition / Competition for Phase II Pathways
- Competition for phase II conjugation pathways (glucuronidation, sulfation) → unconjugated drug is shunted back toward CYP oxidation → apparent CYP inhibition in high-load settings.
- Example: Zidovudine + phenytoin compete for glucuronidation → increased CYP-dependent metabolism → lowers the dose threshold for acetaminophen-induced hepatotoxicity.
— Sleisenger & Fordtran's, p. 1658
Clinical Consequences of Enzyme Inhibition
| Inhibitor | Affected Drug | Consequence |
|---|---|---|
| Ketoconazole, itraconazole | Simvastatin, cyclosporine | Myopathy, nephrotoxicity |
| Clarithromycin | Warfarin | Bleeding risk ↑ |
| Omeprazole (CYP2C19 inhibitor) | Warfarin | ↑ anticoagulant effect |
| Ritonavir (CYP3A4 inhibitor) | Most HIV protease inhibitors | Used therapeutically as "booster" |
| Cimetidine | Theophylline, warfarin, phenytoin | Toxicity of each |
| Grapefruit juice | Nifedipine, felodipine, simvastatin | Exaggerated hypotension, statin toxicity |
Key CYP3A4 Inhibitors
"CRACK" — Clarithromycin, Ritonavir, Azole antifungals, Cimetidine, Ketoconazole (also diltiazem, verapamil, grapefruit juice)
Summary Comparison Table
| Feature | Enzyme Induction | Enzyme Inhibition |
|---|---|---|
| Mechanism | ↑ gene transcription → ↑ enzyme synthesis | Block active site / covalent modification |
| Onset | Days to weeks | Hours to days |
| Effect on drug metabolism | Accelerated ↑ | Slowed ↓ |
| Plasma drug level | Decreased | Increased |
| Clinical result | Reduced efficacy; autoinduction; prodrug activation | Toxicity; ADRs; drug interactions |
| Reversibility | Gradual (days–weeks after stopping inducer) | Reversible (competitive) or permanent (suicide) |
| Key examples (inducers) | Rifampicin, phenobarbital, carbamazepine, St. John's wort | — |
| Key examples (inhibitors) | — | Ketoconazole, clarithromycin, ritonavir, cimetidine |
Clinical Significance (Exam Pointer)
- ~56% of drugs associated with adverse drug reactions (ADRs) are substrates for xenobiotic-metabolizing enzymes.
- ADRs from enzyme interactions cost an estimated $100 billion annually and cause over 100,000 deaths per year in the United States.
- Prior to filing a New Drug Application (NDA), identifying which CYPs metabolize a new drug and whether it is an inducer or inhibitor is now mandatory.
— Goodman & Gilman's The Pharmacological Basis of Therapeutics, pp. 134–136; Lippincott Illustrated Reviews: Pharmacology, p. 60; Dermatology 2-Volume Set 5e, p. 131
This is a shared conversation. Sign in to Orris to start your own chat.