Turner syndrome
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive overview of Turner syndrome drawn from authoritative medical textbooks.
Turner Syndrome
Definition & Epidemiology
Turner syndrome results from complete or partial monosomy of the X chromosome and is characterized by hypogonadism in phenotypic females. It is the most common sex chromosome abnormality in females, affecting approximately 1 in 2,000–3,000 live-born females. Notably, the 45,X karyotype is the single most common cytogenetic abnormality found in spontaneously aborted fetuses, accounting for ~18% of chromosomally abnormal abortions — only about 1% of 45,X embryos survive to birth.
— Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 168
Karyotypes
| Type | Frequency | Details |
|---|---|---|
| Classic 45,X | ~57% | Complete absence of one X chromosome |
| Structural abnormalities | ~14% | Isochromosome Xq [46,X,i(Xq)], ring X [46,X,r(X)], deletions Xp or Xq |
| Mosaics | ~29% | 45,X/46,XX; 45,X/46,XY; 45,X/47,XXX; etc. |
Key point on mosaicism: With sensitive molecular techniques, mosaicism may be detected in up to 75% of cases. Patients with a high proportion of 45,X cells have more severe phenotypes; those with readily detectable 45,X/46,XX mosaicism may appear nearly normal and present only with primary amenorrhea.
Y chromosome sequences: 5–10% of patients carry Y chromosome sequences (full 45,X/46,XY or translocated Y fragments), placing them at significantly higher risk for gonadoblastoma.
— Robbins & Kumar Basic Pathology, p. (block 1)
Clinical Features
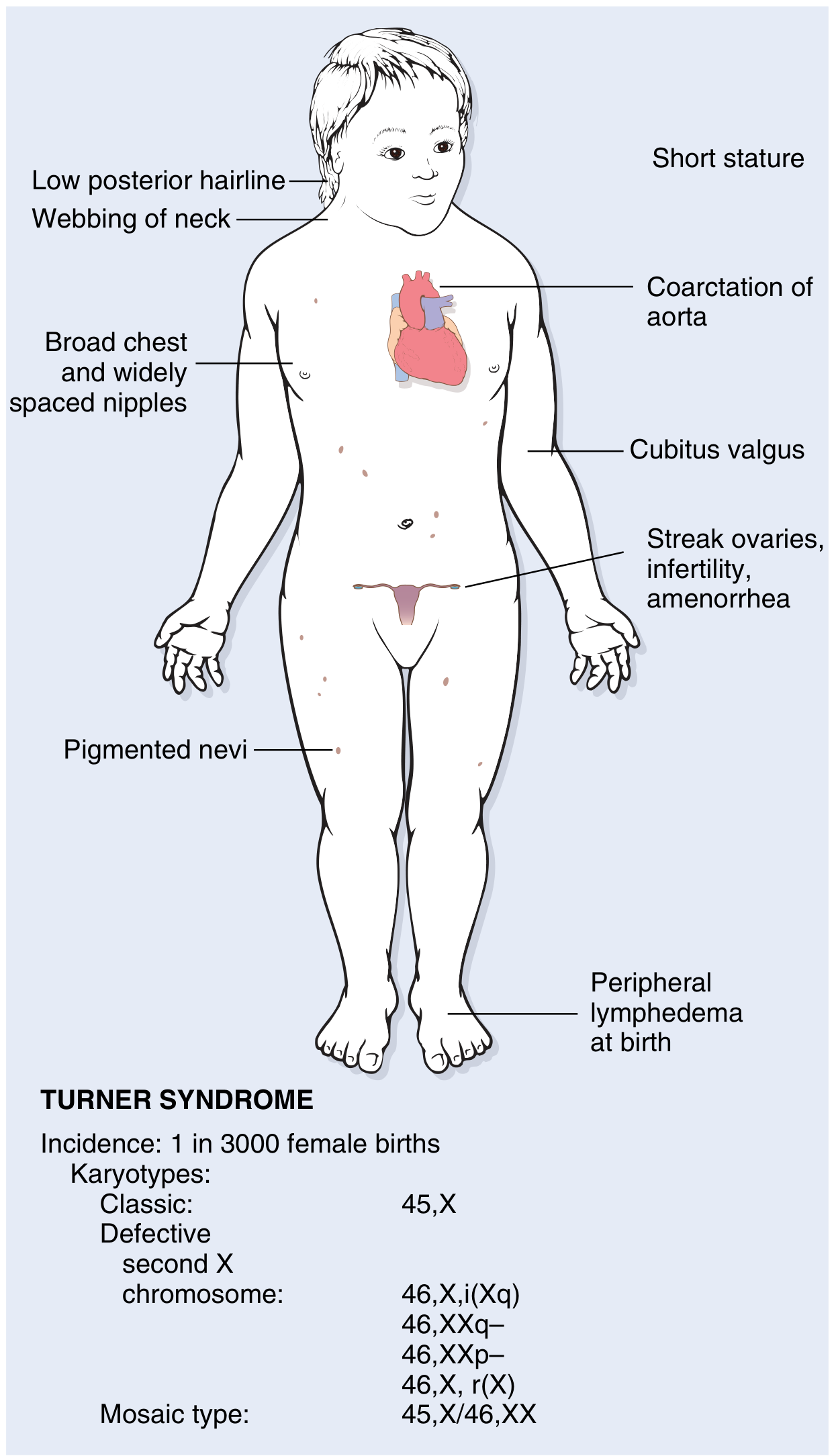
Neonatal / Infantile Presentation
- Peripheral lymphedema of hands and feet (lymph stasis)
- Swelling of the nape of the neck → cystic hygroma → resolves to leave neck webbing and loose posterior neck skin
- Congenital heart disease in 25–50%:
- Preductal coarctation of the aorta (most common)
- Bicuspid aortic valve
- Aortic root dilation (~30%); 100-fold increased risk of aortic dissection
- Cardiovascular disease is the leading cause of death in childhood
Childhood / Adolescent Features
- Short stature (rarely exceeds 150 cm) — below 3rd percentile
- Low posterior hairline
- Webbing of the neck (pterygium colli)
- Cubitus valgus (increased carrying angle of arms)
- Shield-shaped chest with widely spaced nipples
- High-arched palate
- Renal anomalies: horseshoe kidney
- Failure to develop secondary sex characteristics at puberty
- Genitalia remain infantile; minimal breast development; scant pubic hair
- Primary amenorrhea — Turner syndrome accounts for ~one-third of all primary amenorrhea cases
- Pigmented nevi
Reproductive / Endocrine
- Streak ovaries: normal fetal ovarian development up to ~18 weeks, then accelerated oocyte loss — complete by age 2 years. The ovaries are replaced by fibrous strands devoid of follicles and oocytes ("menopause before menarche")
- Infertility in the vast majority; a very small number of mosaic patients can conceive
- ~50% develop autoimmune thyroiditis with clinical hypothyroidism (especially those with isochromosome Xq)
- Subset develops glucose intolerance, obesity, insulin resistance, non-alcoholic fatty liver disease — some fulfill criteria for metabolic syndrome
Cognitive Profile
- Intelligence generally within normal limits
- Subtle deficits in non-verbal, visual-spatial information processing
- Some observations suggest that the parent-of-origin of the single X chromosome influences social adaptation
— Robbins, Cotran & Kumar, p. 168–169
Pathogenesis
- In ~80% of cases the retained X chromosome is maternal in origin — the error occurs in paternal gametogenesis (sperm)
- Key gene: SHOX (short stature homeobox gene) at Xp22.33, located in the pseudoautosomal region — escapes X inactivation, so normally two copies exist in both sexes. Haploinsufficiency of SHOX causes short stature (SHOX deletions are also found in 2–5% of otherwise normal children with short stature)
- Genes on the short arm predominantly determine somatic features; genes on the long arm affect fertility and menstruation
- Loss of one copy of genes in the pseudoautosomal region (shared by X and Y, normally escaping X inactivation) contributes to many features
— Robbins, Cotran & Kumar, p. 169
Diagnosis
- Karyotyping is the gold standard
- Prenatal diagnosis: possible via fetal karyotyping (amniocentesis or CVS)
- Clinical suspicion in adults: combination of short stature + primary amenorrhea
- ~5% of young females diagnosed with coarctation of the aorta have Turner syndrome
Management Highlights
| Issue | Management |
|---|---|
| Short stature | Growth hormone therapy (note: worsens insulin resistance) |
| Hypogonadism / amenorrhea | Estrogen + progesterone hormone replacement therapy |
| Cardiovascular | Regular cardiac imaging; monitor for aortic dilation/dissection |
| Thyroid | Screen for and treat autoimmune hypothyroidism |
| Gonadoblastoma risk (Y+ patients) | Prophylactic gonadectomy |
| Fertility | Assisted reproduction (donor oocytes) in select mosaic patients; VTE risk with exogenous hormones |
— Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar; Berek & Novak's Gynecology; Campbell-Walsh-Wein Urology
This is a shared conversation. Sign in to Orris to start your own chat.