Cardiomyopathy
Reading File
Finding Sources
Searching PubMed
"cardiomyopathy"[MeSH Terms] AND ("classification" OR "management")
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive content from multiple authoritative textbooks. Here is the complete, organized overview:
Cardiomyopathy
Cardiomyopathies are intrinsic diseases of the heart muscle that impair its structure and function, leading to heart failure, arrhythmias, and sudden death. They are classified into three main pathophysiologic categories: dilated (~90%), hypertrophic, and restrictive (least common), plus two additional clinically recognized entities - arrhythmogenic right ventricular cardiomyopathy (ARVC) and Takotsubo cardiomyopathy.
Classification Summary
| Type | EF | Primary Dysfunction | Key Causes |
|---|---|---|---|
| Dilated (DCM) | <40% | Systolic (contractile) | Genetic, viral, alcohol, peripartum |
| Hypertrophic (HCM) | 50-80% | Diastolic (filling) | Genetic (gain-of-function sarcomere mutations) |
| Restrictive (RCM) | Variable | Diastolic (stiff wall) | Amyloid, sarcoidosis, radiation fibrosis |
| ARVC | Variable | RV failure + arrhythmia | Desmosomal gene mutations |
| Takotsubo | Transient | Systolic (apical) | Catecholamine surge, stress |
Source: Robbins & Kumar Basic Pathology, Table 9.5
1. Dilated Cardiomyopathy (DCM)
Definition: Progressive dilation of all four cardiac chambers with contractile (systolic) dysfunction, typically with concurrent hypertrophy. It is the most common form.
Pathogenesis
- Genetic causes (20-50%): Autosomal dominant is most common. Over 50 causative genes identified, primarily encoding cytoskeletal or sarcomere-linking proteins. Titin truncation mutations are the most frequent (up to 20%). Other mutations affect β-myosin heavy chain, α-myosin heavy chain, troponin T, desmin, and nuclear lamins A/C. X-linked DCM is caused by dystrophin mutations (coupling the intracellular cytoskeleton to the ECM).
- Viral infection: Coxsackievirus B, adenovirus, parvovirus B19, human herpesvirus 6. Viral myocarditis can progress to DCM.
- Alcohol/toxic exposure: Alcohol and acetaldehyde are directly cardiotoxic. Also: doxorubicin, cyclophosphamide.
- Peripartum cardiomyopathy: Unique subtype appearing in the last month of pregnancy or up to 5 months postpartum.
- Other: Hemochromatosis (iron overload, Prussian blue-positive myocardium), sarcoidosis, chronic anemia.
Morphology
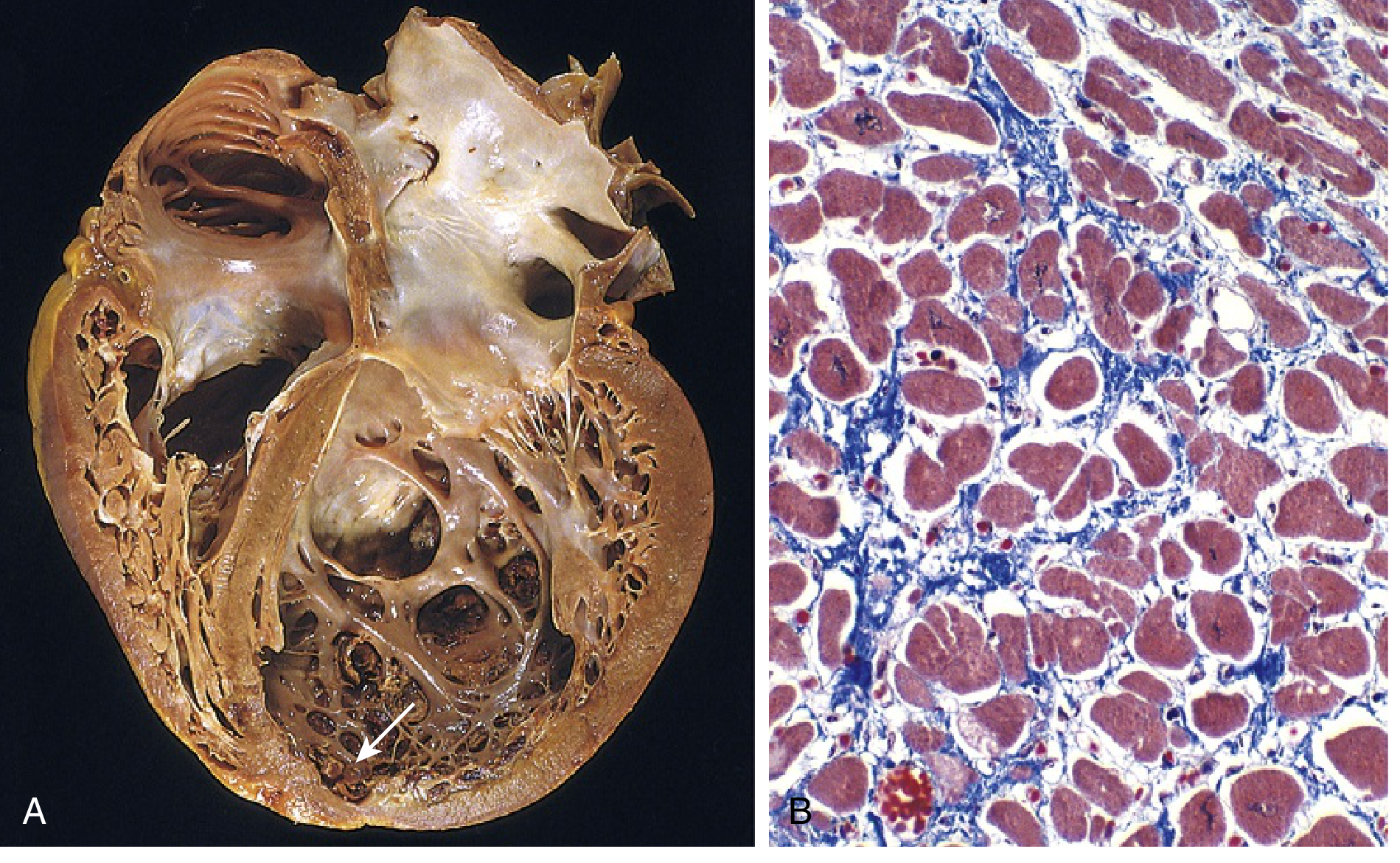
- Heart is enlarged (up to 2-3x normal weight), flabby, with dilation of all chambers.
- Ventricular wall thickness may be normal, thin, or mildly increased.
- Mural thrombi often present (source of systemic emboli).
- Histology: nonspecific - myocyte hypertrophy with enlarged nuclei, attenuated/irregular cells, interstitial fibrosis.
Below: Gross specimen showing four-chamber dilation with apical mural thrombus (arrow, A), and Masson trichrome histology showing myocyte hypertrophy and interstitial fibrosis (B):
Clinical Features
- Most commonly diagnosed between ages 20-50.
- Presents with slowly progressive CHF: dyspnea, easy fatigability, poor exercise tolerance.
- EF typically <25% in end-stage disease.
- Complications: secondary mitral regurgitation, arrhythmias, thromboembolism.
- Median survival to transplant or death: 4-6 years.
- Definitive treatment: cardiac transplantation; long-term ventricular assist devices (LVADs) also used.
2. Hypertrophic Cardiomyopathy (HCM)
Definition: Massive myocardial hypertrophy without ventricular dilation. The heart is thick-walled, heavy, and hypercontractile - the opposite of DCM. Primary dysfunction is diastolic (failure to relax), not systolic.
Pathogenesis
- Almost all cases result from autosomal dominant gain-of-function missense mutations in sarcomeric proteins.
- Over 400 causative mutations in 9 genes. The three most common genes:
- β-myosin heavy chain (most frequent)
- Myosin-binding protein C
- Troponin T
- Together these account for 70-80% of HCM cases.
- Mutations cause myocyte hypercontractility, increased energy use, and net negative energy balance, driving hypertrophy.
- Note: Some of the same genes (e.g., β-myosin) are mutated in both HCM and DCM, but HCM = gain-of-function; DCM = loss-of-function.
Morphology
- Massive myocardial hypertrophy without ventricular dilation.
- In 90% of cases: asymmetric septal hypertrophy - ventricular septum is disproportionately thickened, compressing the LV cavity into a "banana-like" shape.
- Anterior mitral leaflet contacts the septum during systole → systolic anterior motion (SAM) → left ventricular outflow tract (LVOT) obstruction in ~1/3 of cases.
- Histology (characteristic triad): marked myocyte hypertrophy, myofiber/myocyte disarray (haphazard arrangement), and interstitial fibrosis.
Clinical Features
- Typically manifests during the postpubertal growth spurt, but can appear at any age.
- Symptoms: exertional dyspnea (from diastolic dysfunction + LVOT obstruction), harsh systolic ejection murmur, angina (from myocardial ischemia despite normal coronary arteries - due to high LV pressures and compromised intramural blood flow).
- Complications:
- Atrial fibrillation with mural thrombus
- Sudden cardiac death (most feared) - from ventricular fibrillation; HCM is the cause in ~1/3 of sudden cardiac deaths in athletes under 35
- Infective endocarditis (mitral valve)
- CHF
Management
- Medical: Beta-blockers and calcium channel blockers (promote ventricular relaxation).
- Mavacamten: A novel cardiac myosin inhibitor (FDA-approved) that reduces myosin cross-bridge formation, decreasing LVOT obstruction and improving symptoms in obstructive HCM. Studied in the EXPLORER-HCM phase 3 RCT (Lancet, 2020).
- Interventional: Surgical septal myectomy or alcohol septal ablation (controlled infarction of septal muscle via ethanol injection) to relieve outflow obstruction.
- ICD: For high-risk patients to prevent sudden cardiac death.
3. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
Definition: Autosomal dominant disorder marked by right-sided heart failure and life-threatening arrhythmias.
- Prevalence: 1 in 2,000-5,000 adults. Responsible for ~10% of sudden deaths in athletes.
- Pathogenesis: Causative mutations involve genes encoding desmosomal junctional proteins at the intercalated disk (e.g., plakoglobin) and proteins interacting with the desmosome (e.g., desmin). Myocyte death is triggered by desmosomal detachment, especially during strenuous exercise.
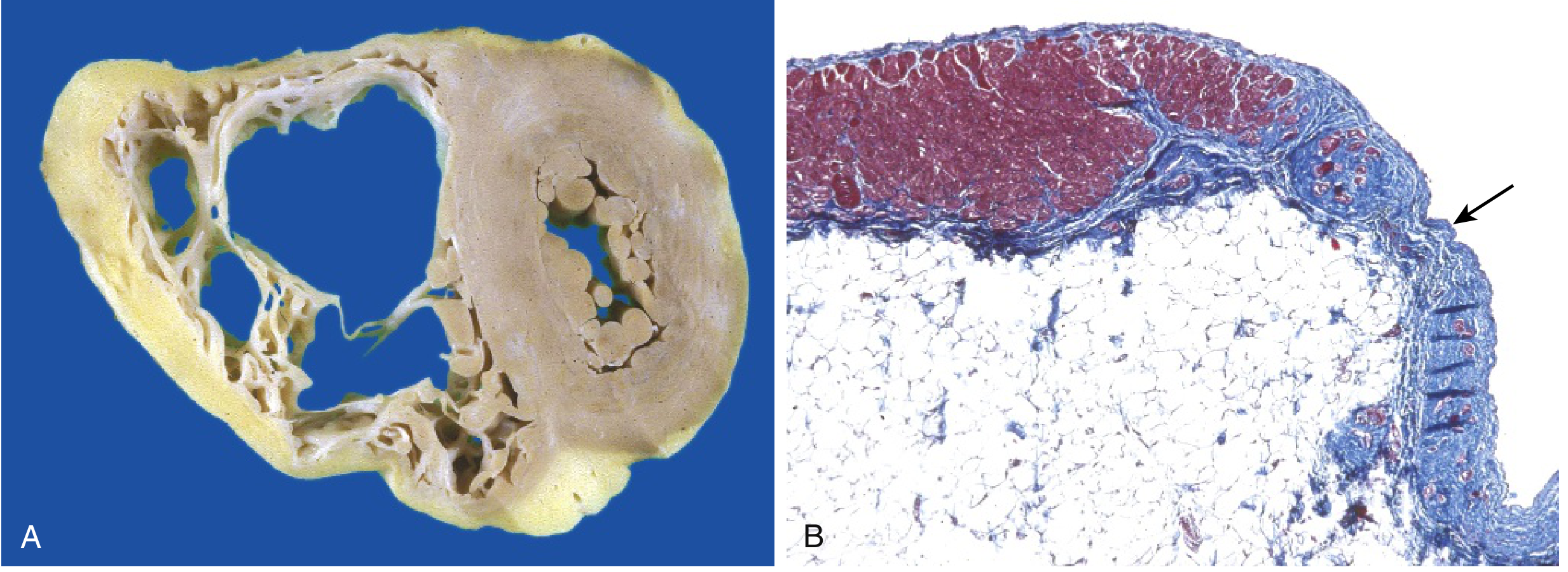
- Morphology: Right ventricular wall is severely thinned due to myocyte replacement by fatty infiltration and fibrosis.
- Clinically: RV failure + ventricular arrhythmias; can cause sudden cardiac death.
Below: Gross specimen showing RV free wall fatty replacement (A) and histology showing myocardium replaced by fibrous tissue (blue, arrow) and fat (B):
4. Restrictive Cardiomyopathy (RCM)
Definition: Decreased ventricular compliance causing impaired filling during diastole - the wall is stiffer. Ventricles are of normal or slightly increased size (not dilated), but both atria are characteristically dilated from elevated filling pressures.
Causes
- Amyloidosis (most clinically important):
- Systemic amyloidosis (AL type, e.g., multiple myeloma) or isolated cardiac amyloidosis.
- Transthyretin (ATTR) amyloidosis: Deposition of normal or mutant transthyretin (a liver protein carrying thyroxine/retinol) in older adults. A specific TTR mutation is carried by ~4% of African Americans, increasing their risk over 4-fold. This is receiving increasing clinical emphasis (Harrison's 22e, 2025).
- AL amyloid light chains are also directly cardiotoxic.
- Endomyocardial fibrosis: Most common RCM worldwide; principally affects children/young adults in Africa and tropical regions. Diffuse fibrosis of ventricular endocardium/subendocardium, often involving AV valves. Associated with nutritional deficiencies and helminthic infections.
- Loeffler endomyocarditis: Peripheral hypereosinophilia + eosinophilic tissue infiltrates; eosinophil major basic protein causes endocardial necrosis → fibrosis → mural thrombus formation.
- Other: Radiation fibrosis, sarcoidosis, mucopolysaccharidoses, sphingolipidoses (Fabry disease), hemochromatosis.
- Genetic: Sarcomere variants causing an RCM-HCM overlap; DES mutations causing combined cardiac and skeletal myopathy.
Diagnosis
- Endomyocardial biopsy often reveals the specific etiology (amyloid deposits, fibrosis pattern).
- Cardiac MRI is the preferred imaging modality.
5. Takotsubo (Stress) Cardiomyopathy
- Also called "broken heart syndrome," "apical ballooning syndrome," or "ampulla cardiomyopathy."
- Epidemiology: Women account for ~90% of cases; prevalence of ~6-7.5% in post-menopausal women presenting with presumed ACS.
- Pathophysiology: Catecholamine surge (physical or emotional stress) causes transient LV apical ballooning with preserved or hyperdynamic basal function. The Fourth Universal Definition of MI classifies this as a separate syndrome, NOT an AMI.
- Prognosis: Often reversible but not always benign - in-hospital mortality 4.1%, long-term mortality 5.6% (International Takotsubo Registry). Recurrence in 5-10%.
- Treatment: No RCTs exist. Beta-blockers do not appear to improve outcomes. ACE inhibitors show some observational benefit (improved 1-year survival). Supportive care is the mainstay.
Myocarditis (Related Entity)
- Inflammatory myocardial damage from infections or immune reactions.
- Most common causes in the US: coxsackievirus A and B, other enteroviruses; increasingly human herpesvirus 6 and parvovirus B19.
- Clinically: ranges from asymptomatic to acute heart failure; can evolve into DCM.
- Giant cell myocarditis, eosinophilic myocarditis, and sarcoidosis are important non-infectious causes.
- Nearly 1 in 5 patients with inflammatory myocarditis harbor pathogenic variants for cardiomyopathy.
Key Genetic Summary
| Cardiomyopathy | Mutation Type | Key Genes |
|---|---|---|
| DCM | Loss of function (cytoskeletal/sarcomere) | Titin, β-MHC, troponin T, dystrophin, desmin, lamins A/C |
| HCM | Gain of function (sarcomere) | β-MHC, myosin-binding protein C, troponin T |
| ARVC | Desmosomal proteins | Plakoglobin, desmin |
| RCM | Sarcomere/TTR variants | Transthyretin (TTR), desmin, sarcomere genes |
Sources: Robbins & Kumar Basic Pathology, pp. 371-374 | Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 2217-2227 | Harrison's Principles of Internal Medicine 22e (2025), p. 2054 | Tintinalli's Emergency Medicine | Braunwald's Heart Disease | Katzung's Basic and Clinical Pharmacology, 16e
This is a shared conversation. Sign in to Orris to start your own chat.