Mechanism of breathing
Reading File
Finding Sources
Finding Sources
Searching the Web
mechanism of breathing physiology inspiration expiration
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive, authoritative content from both Costanzo Physiology and Morgan & Mikhail. Let me compile the full answer.
Mechanism of Breathing
Breathing (pulmonary ventilation) moves air between the atmosphere and the alveoli by generating pressure gradients. The fundamental principle is simple: air flows from high pressure to low pressure. Muscles alter thoracic volume, which changes pressures, which drives airflow.
Key Pressures
| Pressure | Definition | Value at Rest |
|---|---|---|
| Atmospheric pressure | Pressure at the airway opening (mouth/nose) | 0 cm H₂O (reference) |
| Alveolar pressure (Palv) | Pressure inside the alveoli | 0 cm H₂O (= atmospheric; no flow) |
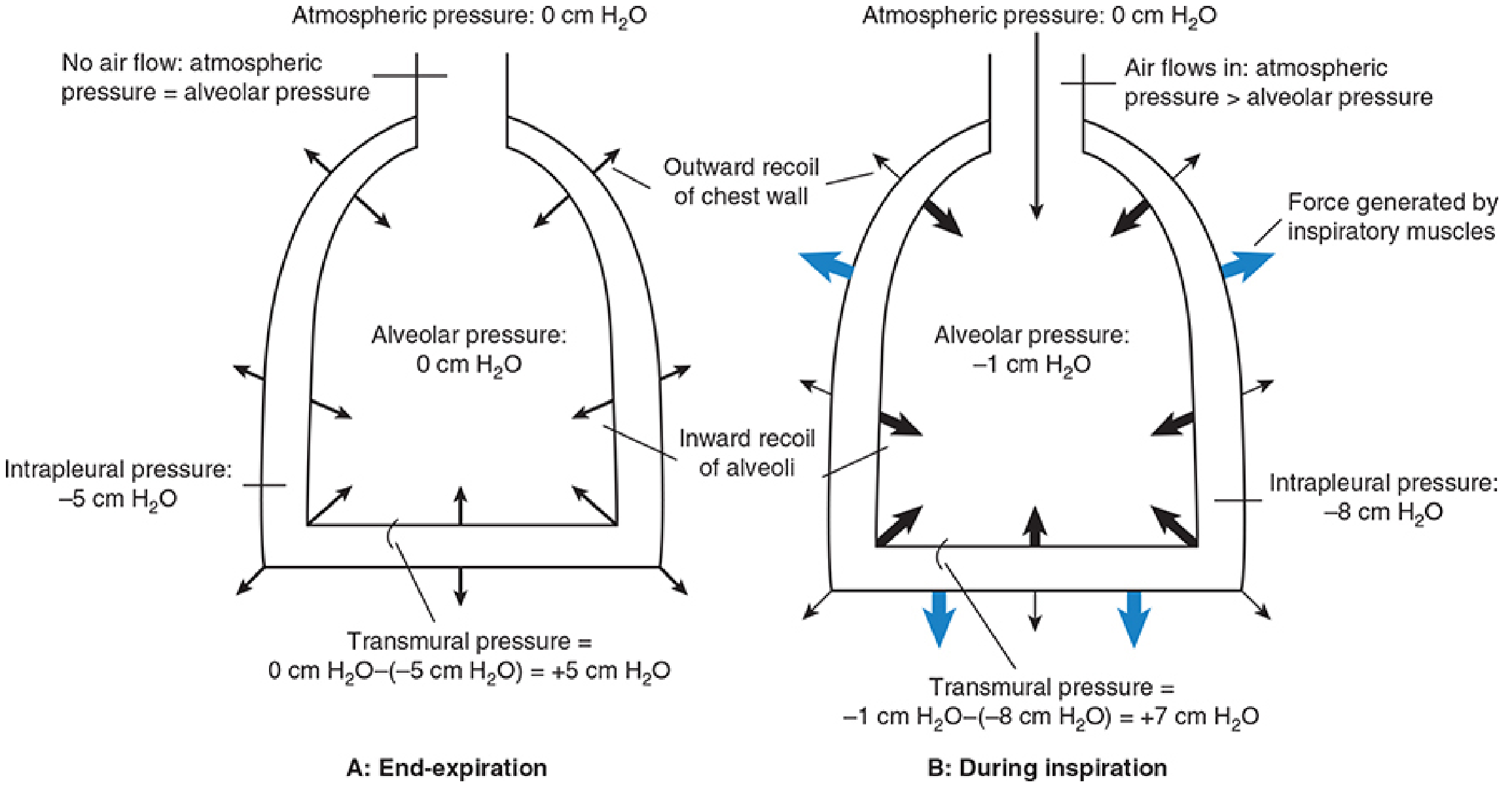
| Intrapleural pressure (Pip) | Pressure in the pleural space between lung and chest wall | -5 cm H₂O |
| Transmural pressure | Palv - Pip; the expanding force across the lung wall | +5 cm H₂O |
Intrapleural pressure is always negative because two opposing elastic forces act across the pleural space: the inward recoil of the lungs (trying to collapse) and the outward recoil of the chest wall (trying to expand). This tug-of-war creates a sub-atmospheric pressure that keeps the lungs inflated at their resting volume - the functional residual capacity (FRC), approximately 2.5 L.
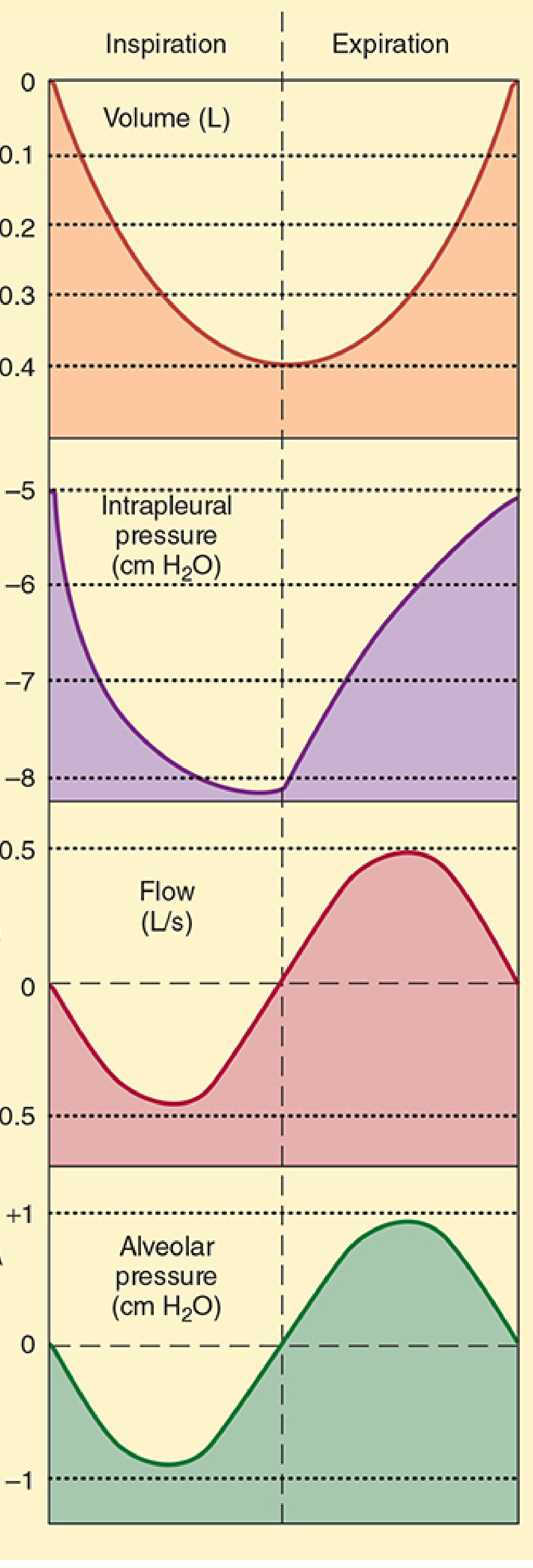
Phase 1: Rest (Between Breaths)
- No air is moving; alveolar pressure = 0 (= atmospheric pressure), so no pressure gradient exists
- Intrapleural pressure = -5 cm H₂O; transmural pressure = +5 cm H₂O (lungs held open)
- Lung volume = FRC
Phase 2: Inspiration (Active)
Inspiration is an active process requiring muscle contraction.
Primary muscles:
- Diaphragm (main muscle) - contracts and descends, increasing thoracic volume vertically
- External intercostal muscles - contract and lift the ribs outward and upward (bucket-handle motion), increasing thoracic volume laterally and anteroposteriorly
Accessory muscles (during exertion or labored breathing):
- Scalene and sternocleidomastoid muscles - elevate upper ribs further
Pressure sequence during inspiration:
- Muscle contraction → thoracic volume increases
- By Boyle's Law (P × V = constant at constant temperature), as volume increases, pressure falls
- Intrapleural pressure drops from -5 to about -8 cm H₂O
- This increase in the transmural pressure gradient expands the alveoli
- Alveolar volume increases → alveolar pressure falls to about -1 cm H₂O
- Now alveolar pressure < atmospheric pressure; air rushes in from the atmosphere
- Air continues to flow in until alveolar pressure returns to 0 (= atmospheric); flow ceases
- The volume of air inhaled in one breath is the tidal volume (~0.5 L); lung volume at end of inspiration = FRC + VT
Phase 3: Expiration (Passive at Rest)
Quiet expiration is entirely passive - no muscle contraction is required.
- Inspiratory muscles relax
- Intrapleural pressure returns toward -5 cm H₂O
- The reduced transmural pressure no longer supports the increased lung volume
- Elastic recoil of the stretched lung tissue compresses the alveoli
- Alveolar pressure rises to about +1 cm H₂O (above atmospheric)
- Air flows out of the alveoli down the pressure gradient until alveolar pressure = 0 again
- Lung volume returns to FRC; the cycle is complete
Forced Expiration (Active)
During exercise, coughing, or deliberate forced breathing, expiration becomes active:
- Internal intercostal muscles and abdominal muscles contract
- They compress the thorax, making intrapleural pressure positive
- Alveolar and airway pressures rise sharply well above atmospheric pressure
- This greatly accelerates airflow out of the lungs
Summary Table: Pressure Changes Through the Breathing Cycle
| Phase | Intrapleural Pressure | Alveolar Pressure | Air Flow | Muscles Active |
|---|---|---|---|---|
| Rest | -5 cm H₂O | 0 | None | None |
| Mid-inspiration | -8 cm H₂O | -1 cm H₂O | Into lungs | Diaphragm, external intercostals |
| End-inspiration | -8 cm H₂O | 0 | None (flow stops) | Diaphragm, external intercostals |
| Mid-expiration (quiet) | -6 cm H₂O | +1 cm H₂O | Out of lungs | None (elastic recoil) |
| End-expiration | -5 cm H₂O | 0 | None | None |
Key Concepts to Remember
- Boyle's Law governs the pressure changes: P₁V₁ = P₂V₂
- Transmural pressure (Palv - Pip) is always positive in normal breathing, keeping the lungs open
- Compliance describes how easily the lungs stretch: C = ΔV/ΔP. Low compliance (stiff lungs, e.g., pulmonary fibrosis) means more work is needed to inflate the lungs
- Airway resistance follows the Hagen-Poiseuille relationship: R = 8ηl/πr⁴. Resistance is inversely proportional to the 4th power of radius - halving airway radius increases resistance 16-fold
- FRC is the equilibrium volume where lung inward recoil = chest wall outward recoil; no muscle energy is needed to maintain it
Sources: Costanzo Physiology 7th Edition, Ch. 5 (Respiratory Physiology) | Morgan and Mikhail's Clinical Anesthesiology, 7e, Ch. 23 (Spontaneous Ventilation) | Tintinalli's Emergency Medicine, Ch. 127 (Respiratory Physiology)
Diff between pharmaco dynamic and kinetic
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Finding Sources
That diagram is perfect - it clearly illustrates the relationship. I now have everything needed to write a comprehensive, well-sourced answer.
Pharmacokinetics vs Pharmacodynamics
The simplest way to remember the difference:
Pharmacokinetics = what the body does to the drug Pharmacodynamics = what the drug does to the body
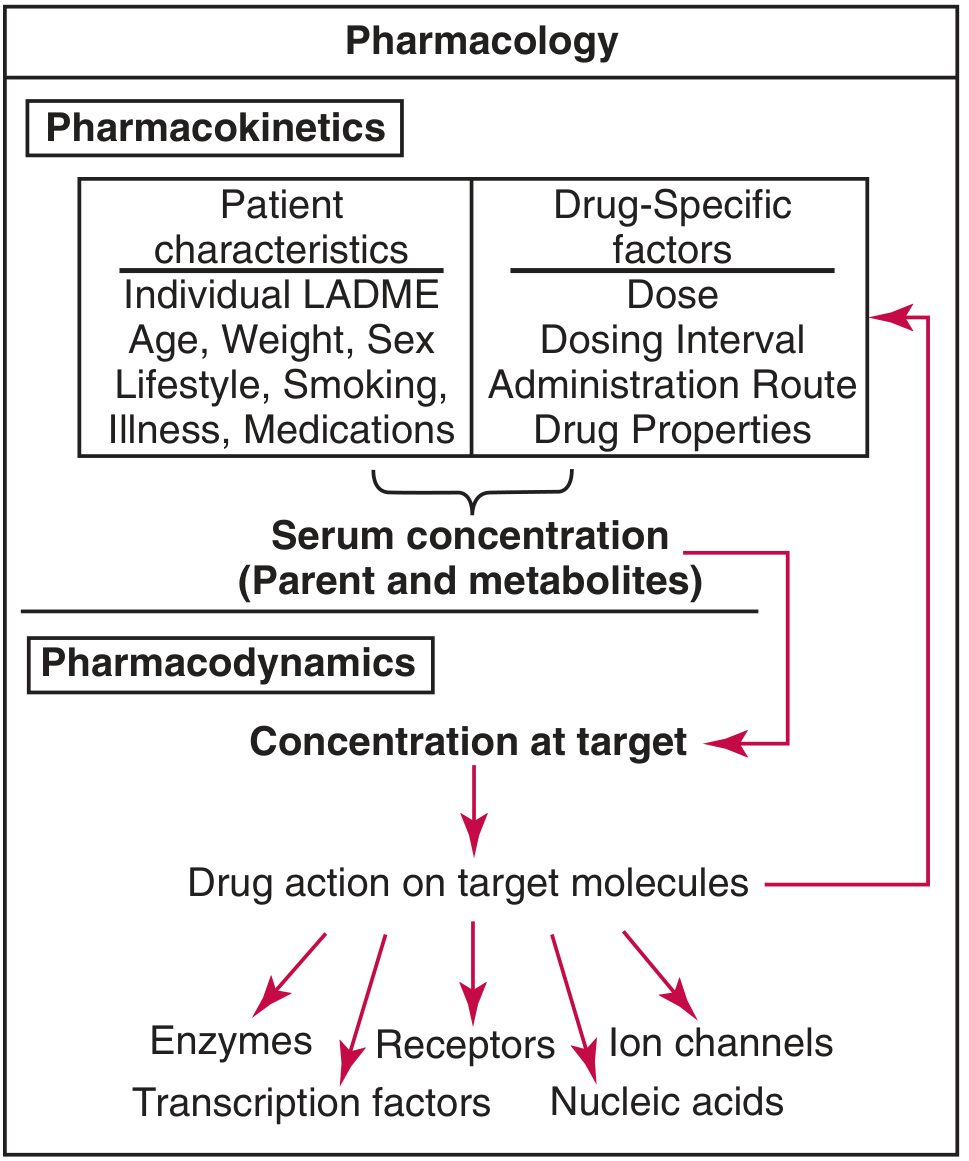
Pharmacokinetics (PK)
Pharmacokinetics describes the movement and fate of a drug through the body over time. It answers: how much drug reaches where, when, and for how long?
The processes are captured in the acronym ADME (or LADME when Liberation is included):
| Step | Process | Key Concepts |
|---|---|---|
| L Liberation | Drug release from formulation | Disintegration, dissolution |
| A Absorption | Entry into systemic circulation | Bioavailability (F), first-pass effect, routes of admin |
| D Distribution | Spread into tissues/fluids | Volume of distribution (Vd), plasma protein binding, blood-brain barrier |
| M Metabolism | Biotransformation, mainly liver | Phase I (CYP450 oxidation/reduction/hydrolysis), Phase II (conjugation), prodrugs |
| E Excretion | Removal from body | Renal clearance (main), biliary, pulmonary; half-life (t½) |
Key PK parameters:
- Bioavailability (F): fraction of administered dose that reaches systemic circulation
- Half-life (t½): time for plasma concentration to fall by 50%; determines dosing interval
- Clearance (CL): volume of plasma cleared of drug per unit time
- Volume of distribution (Vd): apparent volume in which a drug distributes (high Vd = extensive tissue binding)
- First-pass effect: hepatic/gut-wall metabolism before drug reaches systemic circulation (e.g. morphine oral bioavailability ~33%)
Factors affecting PK include age, weight, sex, liver disease, kidney disease, genetics (CYP enzyme polymorphisms), pregnancy, and co-medications.
Pharmacodynamics (PD)
Pharmacodynamics describes the biochemical and physiological effects of the drug and its mechanism of action. It answers: what does the drug do once it gets there, and how intensely?
Core concepts:
- Receptor interaction: most drugs bind to specific receptors (proteins, enzymes, ion channels, nucleic acids, transporters) to produce their effect. The drug-receptor complex must be sufficiently stable to modify the target system and produce an observable pharmacologic response
- Dose-response (concentration-effect) relationship: as drug concentration increases, effect increases - up to a maximal effect (Emax), at which point further increases in concentration produce no additional effect (receptor saturation)
Key PD parameters:
| Parameter | Meaning |
|---|---|
| Emax | Maximum possible effect a drug can produce |
| EC50 (ED50) | Concentration (dose) producing 50% of maximum effect - indicates potency |
| Potency | Amount of drug needed to produce a given effect; inversely related to EC50 |
| Efficacy | Maximum effect achievable; reflects Emax |
| Therapeutic index (TI) | Ratio of toxic dose to effective dose (TD50/ED50); wider TI = safer drug |
| Affinity | How tightly the drug binds to its receptor |
Types of drug-receptor interactions:
- Agonist: binds and activates the receptor (mimics endogenous ligand)
- Antagonist: binds but does not activate; blocks the receptor (competitive or non-competitive)
- Partial agonist: activates but produces less than maximal response even at full receptor occupancy
- Inverse agonist: binds and produces the opposite effect to the agonist
Side-by-Side Comparison
| Feature | Pharmacokinetics | Pharmacodynamics |
|---|---|---|
| Core question | What does the body do to the drug? | What does the drug do to the body? |
| Focus | Drug concentration over time | Drug effect vs. concentration |
| Processes | Absorption, Distribution, Metabolism, Excretion | Receptor binding, signal transduction, physiologic response |
| Parameters | Bioavailability, t½, Vd, CL, Cmax | Emax, EC50, potency, efficacy, TI |
| Clinical use | Determines dosing regimen (dose, frequency, route) | Determines expected effect, selectivity, toxicity |
| Altered by | Age, liver/kidney disease, genetics, drug interactions (enzyme induction/inhibition) | Receptor up/down-regulation, disease states, tolerance, idiosyncrasy |
| Example question | "How much morphine reaches the bloodstream after oral dosing?" | "How strongly does morphine bind to mu-opioid receptors and reduce pain?" |
Why Both Matter Together
PK and PD are inseparable in clinical practice. PK tells you the drug concentration at the receptor; PD tells you what happens at that concentration. The dosage regimen links both: you set the dose and interval (PK) to maintain drug concentrations within the therapeutic window - the range where the drug is effective (PD) but not toxic (PD).
"The overall response to a drug is determined by the interplay of multiple genes involved in both the pharmacokinetic and pharmacodynamic pathways."
Practical example - morphine:
- PK: oral bioavailability ~33% (heavy first-pass metabolism), t½ ~2-3 hours, mainly renal excretion of metabolites - dose must be adjusted in renal failure
- PD: binds mu-opioid receptors → inhibits adenylyl cyclase → decreased neuronal excitability → analgesia, euphoria, respiratory depression; tolerance develops with chronic use (receptor down-regulation)
Sources: Tietz Textbook of Laboratory Medicine 7th Ed., Ch. 42 | Henry's Clinical Diagnosis and Management, Ch. 75 | Katzung's Basic and Clinical Pharmacology 16th Ed., Ch. 3
This is a shared conversation. Sign in to Orris to start your own chat.